![]() (9.16)
(9.16)
CHAPTER IX TESTS FOR PROCESS EVALUATION IN ENVIRONMENTAL ENGINEERING
A. PRECIPITATIVE SOFTENING
Precipitative softening is the most commonly used process for removing hardness cations in drinking water treatment plants. It is based on the precipitation of calcium as calcium carbonate and magnesium as magnesium hydroxide (equation 9.1 & 9.2).
Ca+2 + CO3-2 « CaCO3 ¯ (9.1)
Mg+2 + 2OH- « Mg(OH)2 ¯ (9.2)
Chemical Basis
For this purpose, lime (Ca(OH)2) and soda ash (Na2CO3) are added in proportion to the hardness and alkalinity. This is called lime-soda softening. In order to precipitate calcium, the solubility product constant for calcium carbonate must be exceeded (equation 9.3). For magnesium removal, the solubility product constant for the hydroxide must be exceeded (equation 9.4).
[Ca+2][CO3-2] = 10-8.15 (9.3)
[Mg+2][OH]2 = 10-9.2 (9.4)
Since at the pH of most natural waters, hydroxide and carbonate concentrations are quite low (bicarbonate concentration may be high, however), high concentrations of calcium and magnesium can exist in solution and the concentration products will still be below their respective solubility product constants. In other words, the magnesium and calcium will not precipitate.
If, either the carbonate or hydroxide concentrations can be made to increase, precipitation will occur. By adding lime, naturally-present bicarbonate is converted to carbonate (Equation 9.5). In order for this reaction to occur at a reasonable rate, enough lime must be added to completely react with the bicarbonate present. If manganese is also to be removed, then additional lime must be added to account for the precipitation of manganese hydroxide (Equation 9.6). Experience has shown that more must be added to achieve a sufficiently high pH for rapid precipitation of magnesium. Note that calcium removal results in removal of bicarbonate and carbonate as well.
2 HCO3- + Ca(OH)2 ® 2 CO3-2 + Ca+2 + 2 H2O (9.5)
Mg+2 + Ca(OH)2 ® Mg(OH)2 ¯ + Ca+2 (9.6)
Let us restate the lime softening chemistry. Once all of the bicarbonate is converted to carbonate, addition of lime results in an accumulation of hydroxide and a rise in pH. This excess hydroxide will eventually result in the precipitation of magnesium hydroxide. Therefore, for magnesium removal to approach completion, the required molar dose of lime is equal to one half the bicarbonate concentration plus the magnesium concentration (equation 9.7). In practice, it is necessary to raise the pH to about 11 to accelerate manganese precipitation. This requires the addition of an excess of lime amounting to one-half mM/L.
[Lime Dose] = 0.0005 + [Mg+2] + 0.5*[HCO3] (9.7)
where magnesium, bicarbonate and lime are all in moles/liter.
When the amount of bicarbonate originally-present is insufficient to precipitate all the calcium (originally-present and added), soda ash must be added. The amount of soda ash needed will be equal to the "bicarbonate deficit". This can be calculated from the amount of lime added (according to equation 9.7) plus the calcium originally-present (i.e, [Ca+2]) minus the bicarbonate originally present (i.e., [HCO3]). This is represented in equation 9.8. If the calculated molar dose is less than zero, the system is not bicarbonate limited and soda ash should not be added. In such a case precipitation of carbonate is incomplete, while calcium and magnesium precipitation approach completion.
[Soda Ash Dose] = 0.0005 + [Mg+2] + [Ca+2] - 0.5*[HCO3] (9.8)
Post-precipitation
Precipitative softening is slow chemical process. The precipitation of calcium is a good example of a chemical equilibrium which is kinetically controlled within the time frame of interest (minutes-hours). For this reason, precipitation of hardness cations will continue into the distribution system unless something is done to stop it. Commonly, the pH is lowered by recarbonation (addition of carbon dioxide) or acidification prior to discharge into the distribution system. The desired final pH may be calculated by using the solubility product for calcium carbonate and the acid dissociation of bicarbonate. For example, rearranging equation 9.3, we get:
[CO3-2] = 10-8.15/[Ca+2] (9.9)
Similarly the equilibrium equation for the dissociation of bicarbonate to carbonate can be rearranged to give:
[H+] = 10-10.33[HCO3-]/[CO3-2] (9.10)
Now substituting equation 9.9 into 9.10 gives:
[H+] = 10-2.18[Ca+2][HCO3-] (9.11)
and taking the negative of the logarithm of both sides:
pHs = 2.18 - log[Ca+2] - log[HCO3-] (9.12)
We use the subscript "s" in equation 9.10 to indicate that this is the pH of saturation with respect to calcium carbonate. Above this pH the system is no longer in equilibrium (i.e., the solubility product constant is exceeded) and calcium carbonate should be precipitating. Below this pH, the system is undersaturated with respect to calcium carbonate. In this case calcium carbonate will not precipitate, and in fact, any precipitated calcium carbonate that comes in contact with such a water will tend to dissolve.
Langelier Index
The tendency of a water to precipitate or dissolve calcium carbonate can be characterized numerically by the Langelier Index. This value is defined as the difference between the actual pH (pHa) and the saturation pH (equation 9.13).
L.I. = pHa - pHs (9.13)
If
L.I. > 0, system is oversaturated
L.I. < 0, system is undersaturated
The desired Langelier Index is usually not zero, but rather some small positive number on the order of +0.8 to +1.0. This is due to the tendency of a precipitating water to form a protective coating of calcium carbonate around the inside of iron pipes. This coating will often inhibit corrosion in such systems. However, too much precipitation will lead to clogging of the pipes and reduced hydraulic capacity.
Ionic Strength Effects
The equilibrium constants used to calculate pHs are all based on the ideal case of infinite dilution (m=0). In dilute waters, this is a good approximation, however, waters subject to softening generally have higher ionic strengths. For this reason, corrections must be made on equation 9.12.
All rigorous chemical equilibria are expressed in terms of activity (i.e., {X-}, see Chapter 5). In dilute solution, these activities are nearly equal to the analytical concentrations (i.e., [X-]), and we generally express them as such. However, in waters of high ionic strength, differences between concentration and activities must be taken into account, usually by means of an activity coefficient, g .
{X-} = g x[X-] (9.14)
When activity coefficients are inserted into the equations for the solubility product of CaCO3 and the dissociation of HCO3, and equation 9.12 is re-derived, one gets:
pHs = 2.18 -logg Ca -log[Ca+2] -logg HCO3 -log[HCO3] (9.15)
Activity coefficients may be calculated from the Guntelberg approximation of the Extended Debye-Hückel formula (equation 9.16).
![]() (9.16)
(9.16)
The ionic strength, I, can be estimated from conductivity by the Russell approximation (equation 9.17), or from TDS by the Langelier approximation (equation 9.18).
I = 1.6x10-5 x Specific Conductance (in mmho/cm) (9.17)
I = 2.5x10-5 x TDS (in mg/L) (9.18)
B. COAGULATION JAR TESTING
Coagulation jar testing is a simple laboratory-scale test intended to simulate many of the processes that occur during full-scale coagulation and settling. It is especially useful for selecting optimum coagulant doses, coagulant types and combinations. A variable-speed six-paddle stirrer is used for these tests (see below). Coagulants are added to the "jars" (actually 1000 mL or 600 mL beakers or square plexi-glass containers) during a rapid mix period. This is done with a pipet while the paddles are rotated at their highest speed. Next a period of slow mix is used to simulate flocculation. Finally the paddles are removed from the beakers and the floc is allowed to settle.
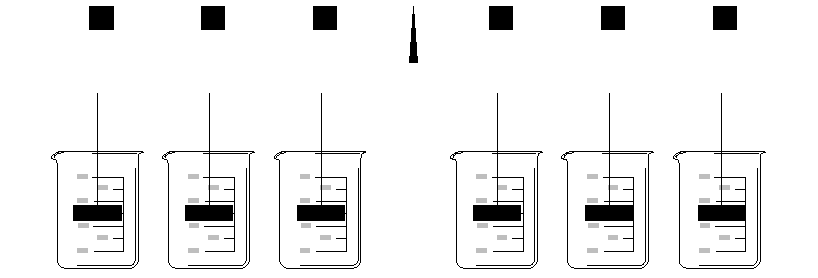
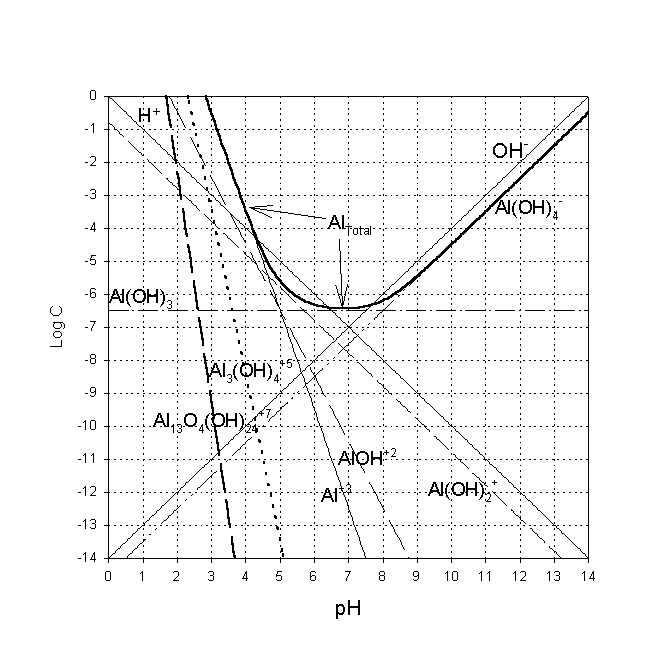
Coagulation processes are defined by the chemistry of the particular coagulants used. In the case of alum (aluminum sulfate), a zone of minimum solubility exists near pH 6 (see graph below). This is the region where the aluminum hydroxide precipitates most rapidly and completely.
C. CHLORINATION BYPRODUCT PRECURSORS
Formation Potentials vs Simulated Distribution System Tests
One can distinguish 3 categories of DBP precursor tests: (1) the high-dose formation potential (HDFP) tests; (2) the low-dose formation potential (LDFP) tests; and (3) the SDS tests. The HDFP is characterized by two attributes (a) it is not used to simulate any particular water system or water treatment scenario; and (b) it uses a sufficiently high chlorine dose so that the residual remains high and nearly constant with contact time, and independent of chlorine demand (e.g., many use a dose of 20 mg/L for samples with a chlorine demand of 8 mg/L or less, so that the residual is always between 12 and 20 mg/L). The HDFP is trying to be an unbias precursor test. This means it exhibits the same percent precursor recovery regardless of precursor concentration. Changing TOC concentrations in waters of high bromide does not present a complication with the HDFP, because the TOC "sees" the same oxidizing environment, regardless of what the actual TOC concentration is. This is not the case for the other tests.
The LDFP is also characterized by two attributes (a) it is not used to simulate any particular water system or water treatment scenario; and (b) it uses a low chlorine dose so that the residual at the end of the contact time is close to what is commonly found in the taps of most US public water systems. The exact chlorine dose is adapted in some way to the sample's chlorine demand. Note that the Uniform Formation Test (Summers, 1993) is a type of LDFP. Tests that fall into this group have an inherent bias toward higher precursor recoveries for more highly colored waters. On the other hand, if carefully run, these tests can provide more accurate information for assessing the expected DBP concentrations at consumer's taps.
The third type of test, the SDS, is characterized by a single attribute: it is designed to simulate the formation in a particular system on a particular day. This test uses a site-specific chlorine dose, pH, temperature and contact time. The values chosen are either based on an existing system on a particular day; or they are based on a very specific scenario intended to simulate a postulated system.
Each of these three types of tests has its own appropriate use. The HDFP is most useful for the assessment of process performance. It can tell you what level of precursor removal is being achieved. The LDFP is most appropriate for comparing finished waters from parallel and alternative treatment trains. It is generally used when the precise disinfection scenario or distribution system characteristics are uncertain. It is not intended for assessing precursor removal across processes, just comparative precursor levels in the finished waters. The SDS is what should be used when the most accurate information about compliance and real-world concentrations are needed. The HDFP is the easiest test to run, and it is the most precise. This is because it is nearly independent of chlorine dose, so errors in dosing or excessive demands will introduce very little error. The LDFP and SDS tests are more labor-intensive, and they are prone to larger uncertainties. In summary, the three types of precursor tests should be used as follows:
· HDFP- for studies of isolated process performance; for understanding the behavior of complex treatment systems; for extrapolating findings at one utility to systems elsewhere in the country
· LDFP- for comparisons of parallel treatment trains at a single pilot plant.
· SDS- for estimating whether a new or midified treatment system will achieve compliance.
Instantaneous DBPs, Terminal DBPs, and Available Precursors
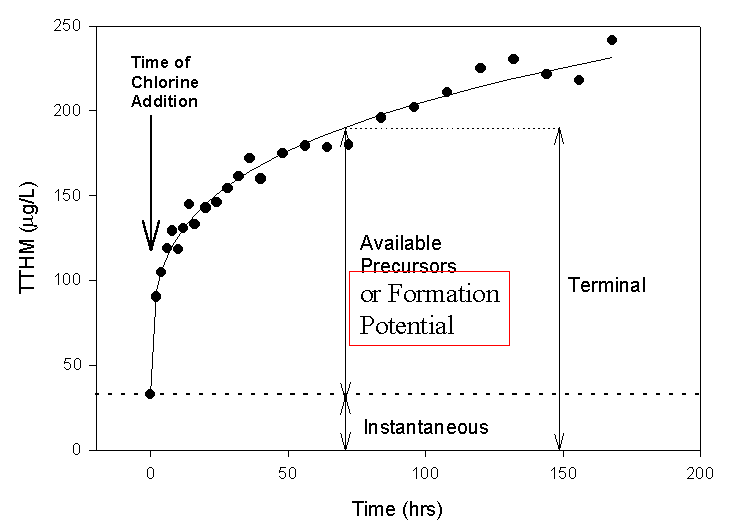
The concentration of trihalomethanes or any other DBP in a sample of water taken from a process stream in a treatment plant or from a tap in a distribution system is referred to as simply the THM concentration or the instantaneous-THM concentration. This is the concentration that existed at the time of sampling. In many cases it is important to know how much effective DBP precursor organics are left in the sample. For this one typically holds the sample without quenching the residual oxidant, and measures the DBP concentration at some later date. The results of this measurement are called the terminal concentration. The exact conditions used (e.g., holding time, pH, additonal chlorine dose, temperature) will depend on the specifics of the study (refer to section 2.a.1). The difference between the terminal concentration and the instantaneous concentration is then the available precursor content. This is the amount of DBP precursors, present at the time of sampling, that will react to form DBPs under the test conditions used.
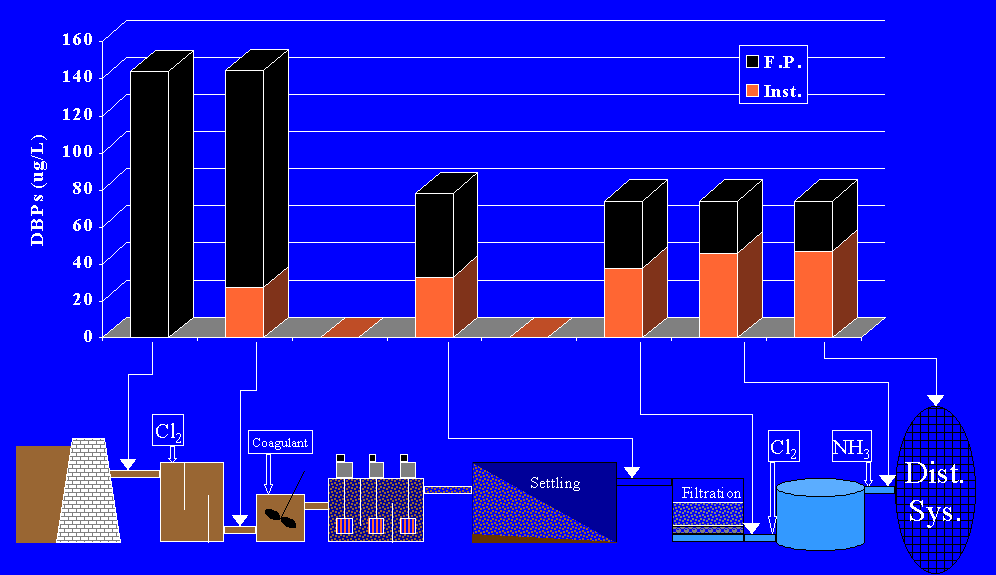
D. BIODEGRADABILITY TESTS
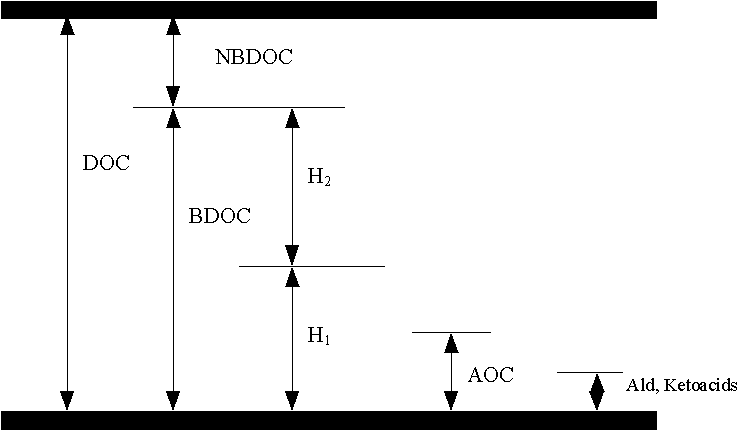
Biodegradable or assimilable organic carbon, depending on the analytical method employed, is defined as either the fraction of dissolved organic carbon (DOC) that can be used by bacteria for growth and cell maintenance or the degree to which microbial growth is stimulated by this DOC. Because of the complexity of natural organic matter, it is impossible to identify the entirety of the DOC. The characterization of biological availability of dissolved organic matter requires the either the use of bioassay techniques or possibly the use of chemical surrogates.
In heavily polluted waters (e.g., municipal wastewater), biochemical oxygen demand (BOD) or chemical oxygen demand (COD) tests are traditionally used for assessing the effectiveness of biological treatment. However, these methods are not sufficiently sensitive for use with treated drinking waters (Rittmann & Huck, 1989). In the past decade, several methods have been proposed to determine the easily assimilable organic carbon (AOC) or the biodegradable dissolved organic carbon (BDOC) in drinking waters. Several of these methods have been review recently by Huck et al. (1990).
1. AOC tests
The first and still the most widely-used method for measuring assimilable organic carbon was developed by van der Kooij et al. (1982). It requires one to follow the growth of a pure culture by a plate count on gelose. The bacteria are inoculated in the water sample, after heating to 60`C. The bacteria commonly used are Pseudomonas fluorescens P17 and Spirillum NOX. The maximum plate count generally achieved after 4 to 8 days of incubation (15`C) is related to the maximum growth of a strain on a specific substrate. It is converted into units of acetate equivalents or oxalate equivalents by referring to a calibration test on a pure acetate or oxalate standard (van der Kooij et al., 1982, Van der Kooij, 1987).
van der Kooij's pure culture approach suffers from several disadvantages. Pure culture of micro-organisms has a more limited capacity for biodegration than a heterogenous bacteria population. Maximum growth counts may be influenced by the physiological state of the Pseudomonas fluorescens culture. The use of pure cultures requires special techniques and highly skilled analysts. The AOC method as it is now commonly used, may also be subject to certain types of bias and large random errors (e.g., LeChevallier et al., 1993). For example, Prévost and co-workers (1992) have noted that difficulties in determining the maximum cell count can lead to significant errors, especially at high AOC concentrations. Possible growth inhibition or delayed growth caused by ozone byproducts and aluminum coagulants has also been reported (Huck et al., 1990). Perhaps these or other factors are responsible for some of the large anomalous increases in AOC-P17 across filtration as noted by Miltner et al. (1992). Nevertheless, the pure culture approach, as exemplified by van der Kooij's, should be subject to less interlaboratory variation, because the innoculum is always the same.
Some laboratories, such as the Water Research Center in England, have modified the method of van der Kooij et al. (1982) by using an inoculum of an autochthonous bacterial population. They have also followed the growth rate through measurements of adenosine triphosphate (ATP) concentration, rather than using plate counts (Jago and Stanfield, 1984).
Rice and co-workers (Reasoner & Rice, 1989; Rice et al., 1990) have developed another pure culture procedure that attempts to measure the ability of coliforms to grow. In this method, a pure culture of coliform (Enterobacter cloacae) is seeded and the ratio of log growth after 5 days (20° C) and time zero is determined. A higher ratio indicates a higher coliform growth response (CGR) and suggests that regrowth of coliforms may be possible in the distribution system. As with many of these alternative bioassays, the correlations with AOC and other tests are poor.
2. BDOC Tests
The methods of van der Kooij et al. (1982), Jago and Stanfield (1984), and Rice (Reasoner & Rice, 1989; Rice et al., 1990) are based on bacterial growth. Other methods have been developed whereby the loss in organic substrate is measured. When the organic substrated is characterized by DOC measurements, the methods are termed biodegradable dissolved organic carbon (BDOC) assays.
Servais and co-workers (Hascoet et al., 1986; Servais et al., 1987; Servais et al., 1989) developed a bioassay that incorporates some aspects of the van der Kooij and Werner methods. The water sample containing dissolved organic matter is sterilized by membrane filtration, inoculated with a second sample of the same water and incubated for 3 weeks at 20`C. After this time the reduction of the dissolved organic carbon (DOC) concentration is measured.
In order to circumvent the long incubation times proscribed by Servais, Joret and co-workers (Joret & Levi, 1986; Joret et al., 1988) proposed that water samples could be tested in the presence of an inoculum of bacteria biomass attached to sand, a more active inoculum, that could reduce the response time to 3 or 4 days.
3. Chemical Surrogates
A very different approach to assessing biodegradability is to use chemical surrogates for AOC or BDOC. As previously discussed, the most widely-used measure of biodegradability in the US is probably the Assimilable Organic Carbon (AOC) assay. Its chief disadvantages are its cost, labor requirements, and long analysis time, and lack of robustness. For these reasons it is not likely to be used as a routine water quality measurement, like dissolved organic carbon (DOC) or trihalomethane formation potential (LeChevallier et al., 1990).
Several researchers have examined alternative chemical assays in order to avoid some of the drawbacks of AOC analysis. Reckhow et al. (1992, 1993) suggested that aldehydes or keto-acids could serve in this capacity. The use of aldehydes has also been supported by Zhou et al. (1992) and Krasner et al. (1993). Both groups of compounds can be measured by gas chromatography following sample extraction and chemical derivatization. Analyses are reproducible, and fast; usually taking only a few hours from start to finish.
The Keto-Acids, and probably the aldehydes are be components of the AOC-NOX, but not the AOC-P17. This is an important characteristic for a proposed surrogate, because these two strains behave very differently. It has been observed that when the two strains are added together, NOX growth is most closely aligned with the concentration of biodegradable organic matter produced by ozonation (e.g., LeChevallier et al., 1992; Shukairy et al., 1992). In contrast, P17 either cannot utilize the ozone byproducts (e.g., oxalate) or it grows more slowly on them, so that its growth more closely reflects the level of naturally-occurring biodegradable organic compounds. Since the precise relationship between AOC levels in finished water and problems related to bacterial regrowth in the distribution system is not known, the desirable level of AOC in drinking waters remains speculative. Many engineers have chosen as a treatment goal the reduction of post-ozonation AOC levels to their pre-treatment levels. This often means that the AOC-NOX produced by ozonation, must be removed by subsequent biological filtration. Therefore, a chemical surrogate specific for AOC-NOX could be especially useful.
Studies conducted by Reckhow et al. (1993) have shown that the keto-acids represent only the most biodegradable of the AOC compounds. Furthermore, there is a fraction of the AOC (even as fraction of the AOC-NOX) that is not readily degraded through water treatment. Similar observations have been made for some of the low molecular weight aldehydes (Miltner et al., 1992; Krasner et al., 1993).
LITERATURE CITED
Crane, G.A. (1988) "Selection of a TOC Analyzer," Am. Lab., July, 1988, pp.51-58.
Edzwald, J.K., W.C. Becker and K.L. Wattier, 1985, "Surrogate Paramters for Monitoring Organic Matter and Trihalomethane Precursors in Water Treatment," J. Am. Wat. Wrks. Assn. 77(4)122-132.
Emery, R.M., E.B. Welch and R.F. Christman (1971) "The Total Organic Carbon Analyzer and its Application to Waterr Research," J. Wat. Poll. Cntr. Assn., 43(9)1834-1844.
Fed. Reg., 1994, US Federal Register, Vol. 59, No. 28, pg. 6332-6444, February 10, 1994.
Handa, N. (1966) "Examination on the Applicability of the Phenol Sulfuric Acid Method on the Determination of Dissolved Carbohydrate in Sea Water, J. of the Oceanographic Soc. of Japan, 22:81-86.
Hascoet, M.C.; Servais, P.; Billen, G. 1986. Use of Biological Analytical Methods to Optimize Ozonation and GAC Filtration in Surface Water Treatment. Proc. AWWA Ann. Conf., pp.205-222., AWWA, Denver, CO.
Huck, P.M.; Fedorak, P.M.; Anderson, W.B. 1990. Methods for Determining Assimilable Organic Carbon and Some Factors Affecting the van der Kooij Method. Ozone:Sci & Eng., In press.
Jago, P. H. and Stanfield, G., 1984. Application of ATP Determination to Measurement of Growth Potential in Water Res. Report, Water Res. Centre, Medmenham.
Joret, J. C. and Levi, Y., 1986. Methode Rapide d'Evaluation du Carbone Eliminable des Eaux par Voie Biologique. Trib. Cebedeau, 510:39:3.
Joret, J.C.; Levi, Y.; Dupin, T.; Gibert, M. 1988. Rapid Method for Estimating Bioeliminable Organic Carbon in Water. Proc. AWWA Ann. Conf., p.1715, AWWA, Denver, CO.
Krasner, S.W., M.J. Sclimenti, and B.M. Coffey, 1993. "Testing Biologically Active Filters for Removing Aldehydes Formed During Ozonation," Jour. Am. Wat. Wrks. Assn. 85:5:62-71.
Le Chevallier, M.W., W.C. Becker, P. Schorr and R.G. Lee, 1992. "Evaluating the Performance of Biologically Active Rapid Filters," Jour. Am. Wat. Wrks. Assn. 84:4:136-146.
LeChevallier, M.W., B.H. Olson, and G.A. McFeters, 1990. "Assessing and Controlling Bacterial Regrowth in Distribution Systems," Am. Wat. Wrks. Assn. Res. Fndtn., Denver, CO.
LeChevallier, M.W., N.E. Shaw, L.A. Kaplan and T.L. Bott, 1993. "Development of a Rapid Assimilable Organic Carbon Method for Water," Appl. Envr. Microbiol. 59:5:1526-1531.
Miltner, R.J., E.W. Rice and R.S. Summers, 1992. "A Pilot-Scale Study of Biological Treatment," Proc. 1992 Am. Wat. Wrks. Assn. Annual Conf., pp.
Pontius, F. W., 1993, "Information Collection Rule to Gather Critical Data," J. Am. Wat. Wrks. Assn. 85(10)16.
Pontius, F.W., 1994, "Information Collection Rule Delayed," J. Am Wat. Wrks. Assn. 86(6)12.
Prevost, M., J. Coallier, J. Mailly, R. Desjardins and D. Duchesne, 1992. "Comparison of Biodegradable Organic Carbon (BOC) Techniques for Process Control," Aqua 41:3:141-150.
Reasoner, D.J.; Rice, E.W., 1989. USEPA Experience with AOC and Coliform Growth Response Assays. Presentation at the Workshop on "Measurement of AOC in the Field of Drinking Water Treatment," Karlsruhe, FRG.
Reckhow, D.A., J.E. Tobiason, M.S. Switzenbaum, R. McEnroe, Y. Xie, Q-W. Zhu, X. Zhou, P. McLaughlin and H.J. Dunn. 1992. "Control of Disinfection Byproducts and AOC by Pre-Ozonation and Biologically-Active In-line Direct Filtration," Proc. 1992 Am. Wat. Wrks. Assn. Annual Conf., pp.487-503.
Reckhow, D.A., Y. Xie, R. McEnroe, P. Byrnes, J.E. Tobiason, and M.S. Switzenbaum. 1993. "The Use of Chemcial Surrogates for Assimilable Organic Carbon." Presented at the 1993 Am Wat. Wrks. Assn. Annual Conf., San Antonio, TX.
Rice, E. W., 1989. Bioassay Procedures for Predicting Coliform Bacteria in Drinking Water. Ph.D. Dissertation, University of Cincinnati, Ohio.
Rice, E.W.; Scarpino, P.V.; Logsdon, G.S.; Reasoner, D.J.; Mason, P.J.; Blannon, J.C., 1990. Bioassay Procedure for Prediction of Coliform Bacterial Growth in Drinking Water. Env. Tech. Let., (in press).
Servais, P., et al., 1987. Determination of the Biodegradable Fraction of Dissolved Organic Matter in Waters. Water Res., 21:4:445.
Servais, P., et al., 1989. Appl. Environ. Microbiol., 55:10:2732.
Shukairy, H.M., R.J. Miltner and R.S. Summers, 1992. "Control of Disinfection By-products and Biodegradable Organic Matter Through Biological Treatment," Revue des Sciences de l'Eau, 5:special:1-15.
Thurman, E.M. (1985) Organic Geochemistry of Natural Waters, Nijhoff & Junk Publ., Boston.
van der Kooij, D., Visser, A., and Hijnen, W. A. M., 1982. Determining the Concentration of Easily Assimilable Organic Carbon in Drinking Water. Jour. AWWA, 74:10:540.
van der Kooij, D., 1987. The Effect of Treatment on Assimilable Organic Carbon in Drinking Water, In Treatment of Drinking Water for Organic Contaminants (Proc., Second National Conference on Drinking Water, Edmonton, Alberta, Canada). Edited by P.M. Huck and P. Toft, Pergamon Press, N.Y., p.317.
Zhou, X.; D.A. Reckhow, and J.E. Tobiason. 1992. "Formation and Removal of Aldehydes in Drinking Water Treatment Processes," Proc. 1992 Am. Wat. Wrks. Assn. Annual Conf., pp.