CHAPTER XIX
GAS CHROMATOGRAPHY
A. INTRODUCTION
Gas chromatography is a powerful technique for separating complex mixtures of organic compounds. It is now the standard technique for the analysis of specific organics in waters and wastewaters. Gas chromatgraphs first became commercially available in 1955. However, it wasn't until the 1970s before they were widely used in the analysis of waters and wastewaters.
The three major components of a gas chromatograph are:
injector
column
detector
Samples are introduced into the flowing gas stream by means of an injector. Separation of the constituents is achieved in the column. Finally, the detector measures the amount of each compound as it leaves the column.
The two major types of gas chromatography are gas-liquid chromatography and gas-solid chromatography. Most environmental applications employ gas-liquid chromatography. This means that the GC column contains a viscous liquid into which the solutes dissolve. This liquid is ultimately responsible for solute separation and peak resolution. In gas-solid chromatography, the columns contain only a solid packing material which serves to adsorb solutes to its surface. In either case a phase change occurs; the former to a liquid, the latter to a solid.
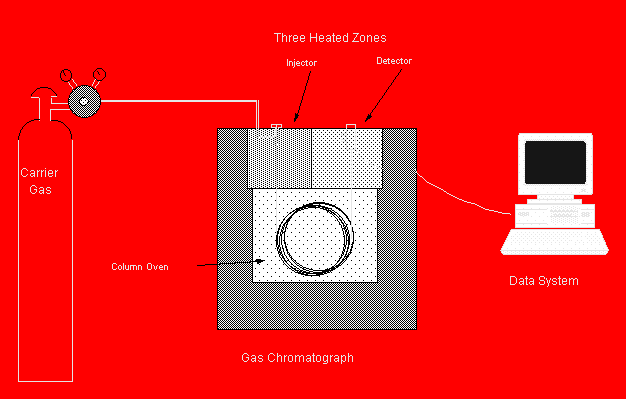
Figure 19.1: A Typical GC System
B.
RATE THEORY OF CHROMATOGRAPHY
This is a simple theory that describes retention time, peak broadening and resolution. As solutes are swept through the column by the carrier gas, they will dissolve to some extent into the stationary phase liquid that is present throughout the length of the column. These solutes will tend to maintain an equilibrium between the stationary phase and the gas phase (carrier gas). This equilibrium is governed by linear partitioning, where the ratio of the concentration of a solute in the stationary phase (Cs) to the concentration in the mobile phase (Cm) is a constant, known as the stationary phase partition coefficient, KS.
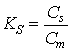 (19.1)
(19.1)
1. Retention Time
The average rate at which a solute migrates along a column, v-bar, is directly proportional to the fraction of time that it spends in the mobile phase.. This is dependent on the partition coefficient.
![]() (19.2)
(19.2)
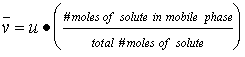 (19.3)
(19.3)
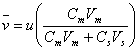 (19.4)
(19.4)
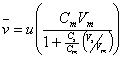 (19.5)
(19.5)
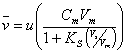 (19.6)
(19.6)
now we define a capacity factor, k'.
![]() (19.7)
(19.7)
so:
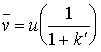 (19.8)
(19.8)
and by definition:
![]() , and
, and ![]() (19.9)
(19.9)
where tm is the residence time of the mobile phase in the column.
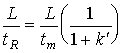 (19.10)
(19.10)
and rearraging:
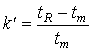 (19.11)
(19.11)
or
![]() (19.12)
(19.12)
The residence time in of the mobile phase may be easily estimated from the retention time of the solvent peak, which is usually unretarded by the stationary phase. Therefore, capacity factors can be determined directly from a chromatogram. The above equations tell us that retention time should be inversely proportional to carrier gas flow rate, and directly proportional to column length, stationary phase thickness and partition coefficient.
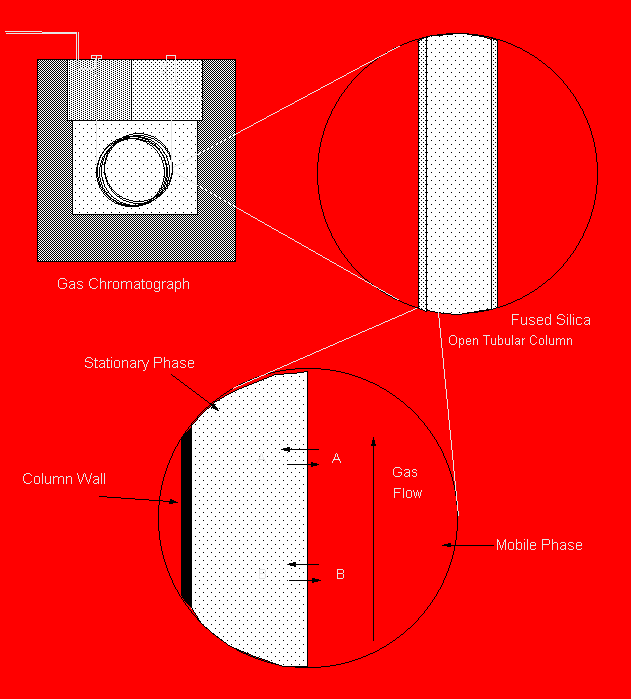
Figure 19.2: Close-up of a Capillary GC Column
2. Peak broadening and resolution
Plate theory holds that the effectiveness of a chrmatographic separation is calculated from:
![]() (19.13)
(19.13)
where:
N is the number of theoretical plates, which is a measure of the overall efficiency of a column
L is the column lenght, and
H is the height equivalent to a theoretical plate (somtimes symbolized as HETP), which is an inverse measure of the column's specific efficiency. The HETP is defined as the length of column needed to achieve a defined degree of solute separation.
The ideal injector deposits its sample in an infinitely small band a the head of the GC column. As the carrier gas sweeps it along, diffusion and dispersion cause the band to spread out. The further it travels the more it spreads out. The ideal concentration distribution of a solute along the length of a GC column traces out a gaussian shaped peak. That is, it has the appearance of a bell-shaped curve. This bell or gaussian curve is characterized by a width parameter, 2, (acutually a normalized variance), a peak height and a position along the column, L. The efficiency of a column (H) is obtained from the peak width divided by the length over which the solute travelled.
![]() (19.14)
(19.14)
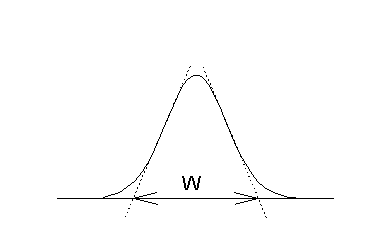
Figure 19.3
A Gaussian Concentration
Profile
And combining these two equations, one gets the number of theoretical plates in the column from the inlet to any point, L, along its length:
![]() (19.15)
(19.15)
In practice one cannot observe the concentration of a solute in a GC column, but with the aid of a detector, one can monitor its concentration as it exits the column. If one draws two lines tangent to a point at half-height on either side of the peak, and extend these lines to the baseline, a line segment which is 4 is marked out.
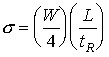 (19.16)
(19.16)
and combining:
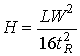 (19.17)
(19.17)
finally,
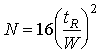 (19.18)
(19.18)
Best resolution or compound separation is obtained with a column having the highest number of theoretical plates, N. In general, the number of theoretical plates a column has increases with:
increasing column length
decreasing stationary phase thickness
decreasing viscosity of the stationary phase
C.
COMPONENTS OF A GAS CHROMATOGRAPH
Temperatures must be carefully controlled at all stages. This is necessary so that the solutes remain in a vapor phase, and so that the temperature-dependent partition coefficients are not allowed to vary uncontrollably.
1.
GC Injectors
The purpose of the injector is to introduce the sample as a single "slug" of vapor. To reach and/or maintain a vapor phase, the temperature must be kept around 50°C or more above the boiling point of the least volatile constituent. When introducing liquid samples, the injector must be capable of rapidlly vaporizing the liquid. Liquid volumes commonly used are:
Packed
columns: 0.2µL to 10µL
Capillary
columns: 0.001µL to 1µL
Flash Vaporization Injector. This is the common type of packed column injector. It typically contains a inert rubber septum that must be pierced by the syringe needle to access the flow stream. The sample enters a small chamber with heated sides. The solvent and constituents rapidly vaporize and the entire flow enters the packed column. Althouth the septum is self-sealing, it may begin to leak after about 25 injections. The septum must than be replaced with a new one, otherwise leaking carrier gas will cause gas flow rates though the column to be reduced, and retention times would shift.
Splitless Injection. This is essentially, the capillary version of a flash-vaporization injector. All of the sample is deposited onto the column. As a result there is no possible sample discrimination or preferential loss of analyte. This is method of injection is especially useful for trace analysis. However, it may suffer severe peak broadening and loss of resolution unless special techniques are employed (e.g., cold trapping, use of solvent effect). Most commonly, a single injector will permit split and splitless operation.
Split Injection. Small sample volumes must be used with capillary GC . If microliter volumes were applied directly to the column as is done with packed-column GC, the vapor slug would fill a very long length of tubing, and extensive peak broadening would result. The best resolution is obtained when sample volumes are as small as possible given sensitivity constraints. However, volumes much less than a few tenths of a microliter cannot be accurately measured or injected. For this reason many GC injectors are equipped with a splitting valve. This redirects most of the carrier gas flow to a vent. By use of this split injector, an easily-measured volume (e.g., 1µL) of sample can be injected and pared down in the injector so that some fraction of this volume (e.g., 0.2%-10%) is actually dellivered to the column. The percent of sample that gets to the column is determined from the split ratio, the ratio of split flow to on-column flow. For example, if the split ratio is 25:1, then 4% of the injected sample will enter the column. Split injection is probably the most common technique for sample introduction. Most split injectors are also capable of splitless injection, and therefore they are called "split-splitless injectors".
2.
GC Columns
Gas chromatographic columns are available in seemingly endless variety. The characteristics that define a column are:
tubing characteristics
- length, inside diameter, composition
stationary phase
- chemical composition, thickness
support
- type, size
treatment
- de-activation
Current practice is to use either capillary or megabore columns. These provide the superior resolution necessary for separating complex environmental mixtures. In the 1970s the standard column was a ¼" ID glass or stainless steel tube that was "packed" with stationary phase coated on a inert granular support. These packed columns were typically 6 feet in length. They contained large granules of inert matierial, called support. The support was almost always a processed diatomaceous earth product. The Chromosorb series of supports were widely used. The purpose of this material was to hold the stationary phase in place and to provide a large surface area for the stationary phase to contact the mobile phase. The support had to be uniform, roughly sherical, and above all, inert. In general, the smaller the better, however if it became too small, pressure loss would become unacceptable. Common sizes include 60/80 mesh and 80/100 mesh. If the silica surface on the support or on the wall of the glass column become hydrated they will form silanol groups. These can hydrogen bond with polar and polarizable constituents in a sample and cause a deterioration in the chromatographic resolution. Peaks will have long tails that interfer with subsequent peaks, rather than ending sharply. These active surfaces can be de-activated with dimethylchlorosilane (DMCS) and methanol as shown below.
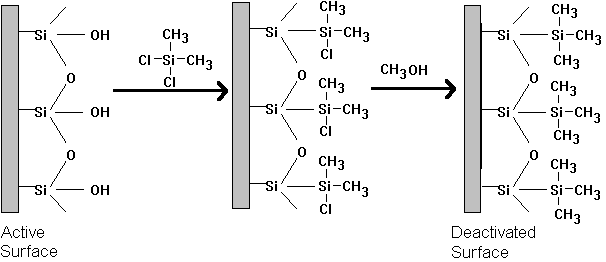
Figure 19.3: Column Inactiviation
Acid washing can also be useful for removing mineral impurities. Columns that have been so treated are designated with the suffix, "-AW-DMCS". If instead, hexamethyldisilizane is used, the designation is "-HMDS".
Although packed columns have far fewer theoretical plates than modern capillary columns, they do offer some advantages. They can accomodate larger samples, and therefore they may improve sensitivity. They are also far easier to use, and the associated injectors are simpler. For these reasons, and because they are less costly, packed columns are still used today for analyses that don't demand high resolution.
Table 19.1
Comparison of Packed and
Capillary Columns
|
Column Type |
Typical Characteristics |
|||
|
|
Inside Dia. (mm) |
Length (m) |
plates/meter |
total plates |
|
Packed |
2-4 |
2-3 |
750-3000 |
1500-9000 |
|
FSOT |
0.1-0.3 |
15-60 |
2000-4000 |
30,000-240,000 |
Liquid stationary phases should have the following properties:
low volatility
thermal stability
chemical inertness
interactions with solutes to give a good separation
Although hundreds of stationary phases exist, there are probably only a dozen or two substantially different types. Most analyses use the methyl or phenyl-methyl silicone phases. These are sold commercially under a variety of names. For capillary columns, they can be bonded to the silica column. Full methyl silicone, or more properly poly(diemthylsiloxane), are given the designation "X"-1, where "X" is the vendor's code name (e.g., HP for Hewlett Packard, DB for J&W Scientific, SPB for Supelco). The 5% phenyl-methyl silicone, or poly(5%-diphenyl-95%-dimethylsiloxane) is designated "X"-5. The methyl silicone is very non-polar. It tends to separate compounds only on the basis of boiling point. The phenyl-methyl silicone provides some selectivity for aromatic compounds, and those that have an affinity for the aromatic bands. Such constituents will be retained a bit longer than they would be with a methyl silicone phase.
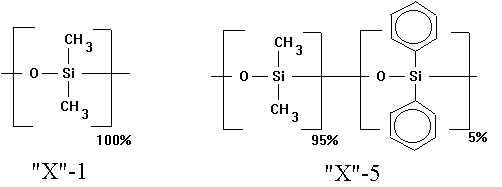
Figure 19.4: Two Common Stationary Phases
In 1979, the fused silica open tubular columns (FSOT) were introduced into the market. These are composed of fused silica surrounded by a polyimide coating. This produces an inert, thermally-stable, yet durable tube that is sold in lengths of 100 m or more. It is fully flexible, and almost unbreakable provided that the brownish imide coating remains intact. Most modern capillary GC applications use FSOT columns with some chemically-bonded stationary phase. The process of chemically bonding the phase produces a uniform thin layer of exceptional thermal stability.
Carrier Gases
Carrier gases should be chemically inert and compatable with the detector used. Most commonly used are Helium, Argon, Nitrogen and Hydrogen. Hydrogen often gives the best performance, but is not often used for safety reasons.
3.
GC Detectors
Thermal Conductivity. The thermal conductivity detector or TCD is one of the earliest and most versatile of the detectors. It responds to change in thermal conductivity as the carrier gas is diluted by organic solutes. The thermal conductivity of the column effluent is acutally compared on a continuous basis with that of a reference flow. This detector requires that either helium or hyrdrogen be used. Beause these gases have such high diffusivities, they also have higher thermal conductivities than nearly all organic solutes. This detector is simple, rugged and exhibits good linearity (over 5 orders of magnitude). Its chief disadvantage is poor sensitivity (~10-8 g/mL). For this reason it cannot be used very effectively with capillary columns which cannot handle large sample volumes.
Flame Ionization. The flame ionization detector or FID measured the ions collected after pyrolysis in a hydrogen/air flame. For this reason, the FID requires that high purity hydrogen and air be available in addition to whatever carrier gas is used. A tiny flame is ignited during the startup of a run and as carrier gas passes out of the column, the organic constituents are converted into ionic intermediates. These charged species are captured by a collector set at 200-300 volts. The number of ions is roughtly proportional to the number of reduced carbons in the column effluent. Carbons of a higher oxidation state (e.g., carboxyl groups, alcohols, ketones, alkyl halides) produce fewer ions and therefore a smaller signal. For this reason, the detector does not respond at all to fully oxidized compounds such as water, carbon dioxide, sulfur dioxide, etc. The FID has an exceptional linear range (7 orders of magnitude), fast response time (due to high hydrogen and air flows), and it is quite sensitive (~10-13 g/mL). Its chief disadvantes are that it is destructive and it requires additional gases. Nevertheless, the FID is one of the most widely-used detectors today.
Thermionic, Nitrogen-phosphorus, or Alkali-flame. The thermionic detector, Nitrogen-phosphorus detector (NPD) and alkali-flame detector are three names for the same detector. It is identical to the FID except for the presence of a rubidium silicate bead near the collector. This "active element" served to dramatically increase the detector's sensitivity for nitrogen and phosphorus compounds. When tuned properly (correct hydrogen and air flows, and correct voltage) the detector is 3-5 orders of magnitude more sensitive for nitrogen than carbon, and 4-6 orders of magnitude more sensitive for phosphorus than carbon. Its sensitivity for these elements compared to the FID is 50 times greater for nitrogen and 500 times greater for phosphorus. For this reason it has found special use for phosphorus-containing pesticides.
Photoionization. The photoionization detector or PID is a general detector which was introduced in 1976. It relies on the use of ultraviolet light to produce ions from organic solutes (equation 19.19). The electrons given off are then measured by a sensitive electrometer. This detector has a sensitivity that is 10-50 times that of an FID. It is essentially non-destructive, and it can be used as a stand-alone instrument for detecting total organic vapors (e.g., HNU "PI-101" gas analyzer, Photovac "Snapshot" vapor analyzer), or in rugged field GCs (e.g., Photovac "10S plus" GC).
![]() (19.19)
(19.19)
Table 19.3
Relativity Sensitivity of
the Photoionization Detector to Selected Organic Compounds
|
Compound |
Sensitivity |
|
p-Xylene |
11.4 |
|
m-Xylene |
11.2 |
|
Benzene |
10.0 |
|
Toluene |
10.0 |
|
Styrene |
9.7 |
|
Trichloroethylene |
8.9 |
|
Iso-butane |
7.0 |
|
Cyclohexanone |
5.1 |
|
Vinyl Chloride |
5.0 |
Electron Capture. The electron capture detector or ECD is widely used for halogenated organic compounds. It contains a small piece of radioactive foil contining 63Ni, a beta emmitter (sometimes 3H is used). These high-energy electrons will ionize the carrier gas, and produce low-energy electrons and positive ions. The secondary electrons are collected at the anode, which is typically operated in a pulsed mode to avoid polarization and collection of large anionic fragments. When an electrophilic constituent passes out of the column and into the ECD, it will pick up some of these low-energy electrons and thereby reduced the anode current. The more electronegative a compound is, the more sensitive the ECD is to it. The most strongly recorded compounds are the halogenated organics, peroxides and nitro-organics. The ECD shows almost no response to amines, alcohols and hydrocarbons. This detector is exceptionally sensitive to halogenated pesticides and chlorination byproducts. It is also highly selective, which means it will not be troubled by interference from non-halogenated compounds. It will, however, respond to oxygen, so care must be taken to avoid any oxygen contamination of the carrier flow.
D. DATA ANALYSIS
1. Qualitative Analysis
The identification of peaks in gas chromatography is a the most important aspect of data analysis. Without some supplemental knowledge about the sample being analyzed, a single gas chromatogram is nothing more than an ill-defined "fingerprint". Fortunately, we often know quite a bit about our samples even before they are analyzed. For example, contaminated groundwater from partially-characterized sites may contain a constituent of a known mixture of petroleum products. Chlorinated drinking water likely contains certain well-defined chlorinated methanes and acetic acids. This information is valuable in coming to a conclusion about the identity of peaks in a chromatogram with an accepted level of certainty.
There are may levels of assurance that a peak identification is accurate. The level which is considered acceptable depends on the nature of the sample and the intended use of the data. The highest levels of certainty require other sophisticated instrumentation besides just GC (e.g., mass spectrometry, infrared spectroscopy). Probably the most certain identification is obtained when a mass spectrometer is used as a GC detector (this combination is called GC/MS), and commercially-available standards are run with the sample. If the mass spectrum produced by the mass spectrometer and the retention time in the GC match between known standard and the peak of interest, then the identification is said to be "MS-confirmed". If a known standard is not available, but the mass spectrum is deduced by logical chemical reasoning to be that compound, the identification is said to be "MS-tentative".
When a qualitative detector such as a mass spectrometer is not available, confirmation of peak identities can be achieved by using two different GC columns or two different quantitative detectors. The co-elution of an unknown peak and a known standard on two different GC columns having different retention characteristics is generally considered sufficient to be designated "GC-verified". The certainty of this identification is increased the higher the column resolution. Another method is to use two different detectors with the same column. If the selectivities of the two detectors are substantially different, then all one needs to do is match the retention time and the ratio of the response from the two detectors. If these can be matched between unknown peak and commerical standard, then the identification is also "GC-confirmed".
The confirmation of peak identities is very important in the analysis of new types of samples or samples from new sites. However, much of environmental trace analysis involves the processing of similar types of samples that only differ in concentration. These may be almost identical in a qualitative sense. It is generally accepted that with such samples, the confirmation of peak identities is not necessary. Rather all one has to do is to conduct a standard addition analysis. Here known amounts of the analytical standard are added to aliquots of a sample and analyzed by GC. If the suspected peak grows with increasing addition of standard, and no new peaks appear, then the qualitative criteria are satisfied. Exceptions include extremely low-level contaminants of great environmental significance, such as chlorinated dibenzo-dioxanes or the disinfection byproduct, "MX". These are so easily obscured by interfering compounds that the presence of peaks which co-elute with known standards must always be view with skepticism.
2. Quantitative Analysis
Quantitative GC analysis is generally based on a presumption of a linear relationship between concentration and peak area or peak height. Before the advent of integrators, peak height was most commonly used. A series of standards of known concentration was run and the response was compared with that of the unknown. Now it is more common to use peak area (Figure 19.5). In principle, either should work as long as the response is ideal. However, changes in retention time from one run to the next will change the height and width of the peaks, but not the area. For this reason, the use of peak area for quantification is probably more robust.
The use of internal standards can dampen errors due to such factors as variable injection volume or split ratio. The internal standard is a compound that is added to the extracts of samples and standards alike. It can be separately quantified from the analytes and it serves as a relative measure of the amount of original extract that was ultimately delivered to the detector.
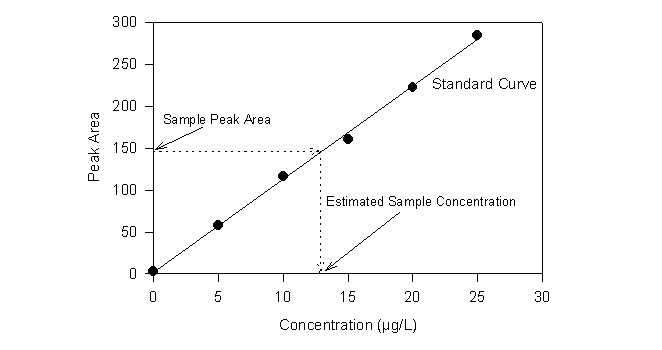
Figure 19.5
Quantitative GC Analysis
with External Standards
E.
GAS CHROMATOGRAPHIC APPLICATIONS
1.
Analysis of Petroleum Hydrocarbons
Gas chromatography offers a very powerful compound-specific alternative to the infrared methods for hydrocarbon analysis. Typically a purge & trap pretreatment is used in conjunction with a methy-phenyl silicone stationary phase and a flame or photo ionization detector. When new or unfamiliar samples are run, a second column containing a different stationary phase is used for confirmation of peak identifications (EPA Method 602).
Example:
Route 128 service station in Lexington, MA
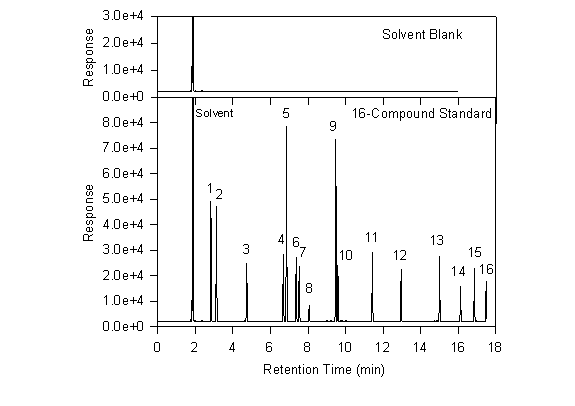
As an example, chromatograms are shown below from a study of contaminated groundwater at Lexington, MA (Ostendorf et al., 1994). Analysis of a sample of the separate-phase floating product revealed dozens of identifiable hydrocarbons. Of these, sixteen major constituents were chosen for use as a calibrating standard. The chromatogram below shows one of these standards along with a solvent blank. The standard is prepared in methylene chloride and a sample of the same methylene chloride is used for the solvent blank. As is usually the case, the solvent peak is the first major one. There are, however, a few tiny nearby peaks that are undoubtedly contaminants in the solvent. These are so small and close to the solvent as to have little or no effect on the analysis of samples. The solvent and its accompanying small peaks will appear in all standards and samples where methylene chloride was used.
By comparing the solvent chromatogram with that from the calibrating standard, one can deduce which new features are due to the presence of the 16 standard compounds. As one would hope, exactly 16 new peaks appear in this chromatogram. By injecting single compound standards one at a time, the identity of all 16 can be determined. They are shown in Table 19.2.
Table 19.2
Calibration Standard used
for Lexington Study
(Ostendorf et al., 1994)
|
Number |
Name |
Retention Time (min) |
|
1 |
2-Methylhexane |
2.83 |
|
2 |
2,2,4-Trimethylpentane |
3.12 |
|
3 |
3-Methylheptane |
4.74 |
|
4 |
Ethylbenzen |
6.69 |
|
5 |
m-Xylene |
6.86 |
|
6 |
o-Xylene |
7.38 |
|
7 |
Nonane |
7.54 |
|
8 |
Cumene |
8.07 |
|
9 |
1,2,4-Trimethylbenzene |
9.48 |
|
10 |
Decane |
9.59 |
|
11 |
Undecane |
11.42 |
|
12 |
Naphthalene |
12.96 |
|
13 |
1-Methylnaphthalene |
15.00 |
|
14 |
Tetradecane |
16.12 |
|
15 |
2,3-Dimethylnaphthalene |
16.85 |
|
16 |
Pentadecane |
17.48 |
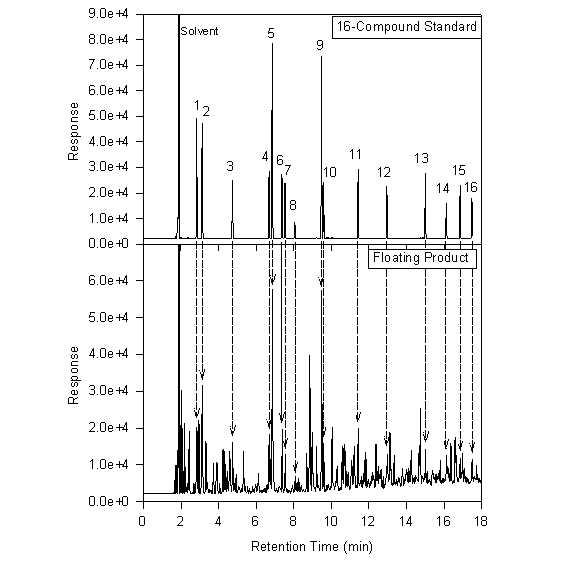
The chromatograms below provide a comparison between the 16-compound standard and an actual separate phase sample. The nature of this petroleum sample is not entirely certain, but it may contain weathered Diesel fuel, gasoline, kerosene and heating oil.
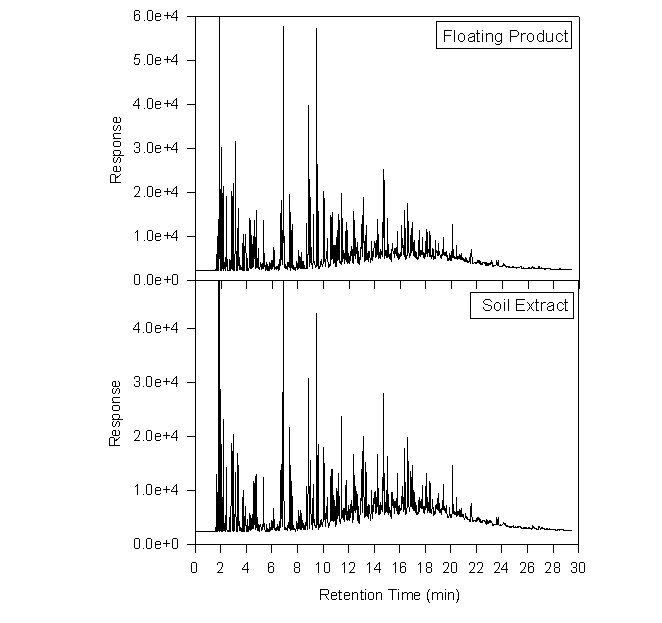
A methylene chloride extract is compared with the separate phase product below. This particular extract came from a soil sample collected on 18 January 1995 at a depth of 114-132 inches. It represents a highly contaminated zone, with a TLV reading of 4200. The two chromatograms are remarkably similar. Note the slightly higher relative abundance of the later eluting peaks in the soil extract, and the higher abundance of the earlier peaks in the floating product. This points to preferential loss of volatiles.
2. Analysis of disinfection
Byproducts
A) Neutral Extractables
LLE,GC,ECD: USEPA #551
P&T,GC, ElCD: USEPA #502.2
P&T,GC,MS: USEPA#524.2
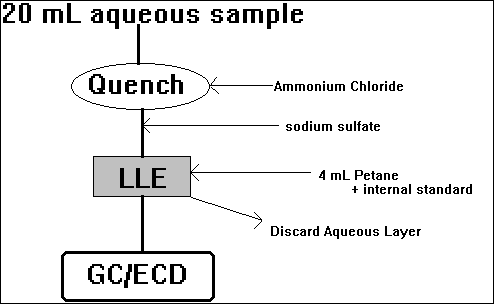
B) Haloacetic Acids
LLE,GC,ECD: Standard Methods, #6233 - the preferred method, but uses diazomethane
SPE,GC,ECD: USEPA #552.1 - acidic methanol, but some recoveries are not good
(spontaneous methylation in methanol stocks)
C) TOX
Activated Carbon Adsorption
&
Pyrolysis
&
Microcoulometric Detection of halide
D) MX
e.g., Charles et al. [1992, Environ. Sci. Technol., 26:5:1030-1035]
Acidification, Extraction
with diethyl ether, KD concentration, BF3/methanol, GC/high
resolution MS
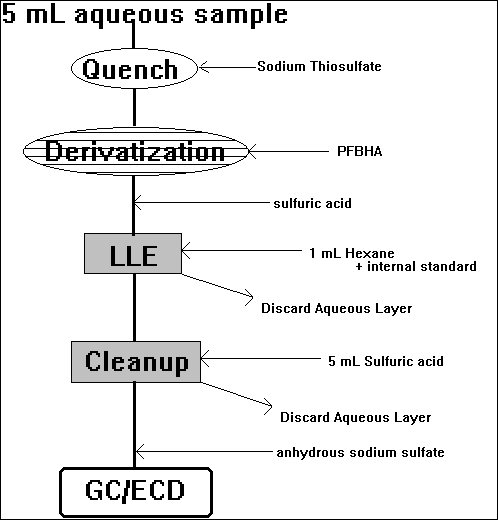
E) Aldehydes
Method of Yamada & Somiya [1989, Ozone: Sci Eng. 11:125-141] as modified by Glaze et al. [1989, Environ. Sci. Technol., 23:7:838-847], and later by Sclimenti et al. [1990, Proc. AWWA WQTC, pp.447-501].
(formation of oximes with PFBHA)
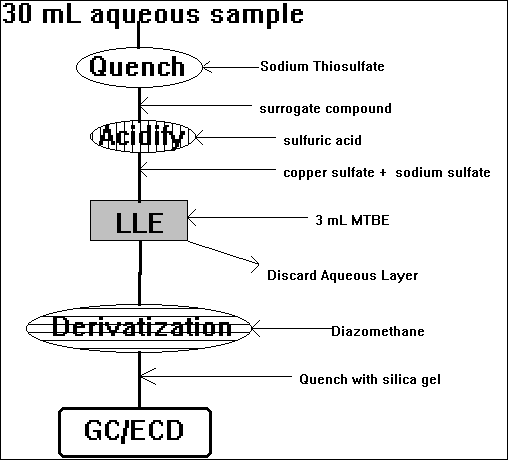
F) Bromohydrins
Cavanagh et al. [1992, Environ. Sci. Technol., 26:8:1658-1662]
mostly analytical artifacts?
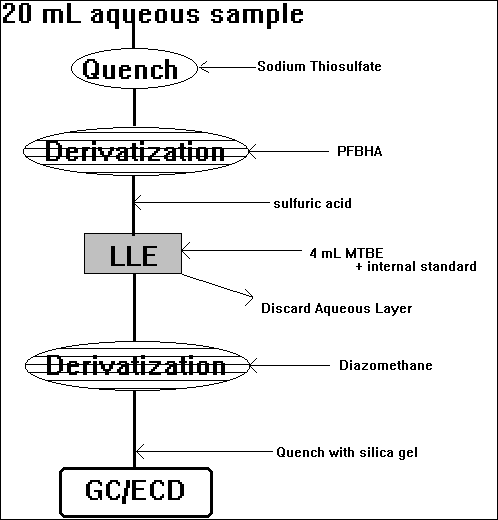
G) Ozonation Byproducts
Aldehydes
Ketoacids
(double derivatization)
Literature Cited
Ostendorf, D.W., A.J. Lutenegger, D.J. DeGroot, E.S. Hunt, P.S. Cheever, E.S. Hinlein, R. Suchana, and J. Jordan (1994) "Lateral Distribution of Petroleum Hydrocarbons at a Leaking Storage Tank Site," Interim Report, prepared for the Massachusetts Highway Department.