CHAPTER XV: GRAVIMETRIC METHODS
A. INTRODUCTION
1. Definitions
All Gravimetric analyses rely on some final determination of weight as a means of quantifying an analyte. Since weight can be measured with greater accuracy than almost any other fundamental property, gravimetric analysis is potentially one of the most accurate classes of analytical methods available. These methods are among the oldest of analytical techniques, and they may be lengthy and tedious. Samples may have to be extensively treated to remove interfering substances. As a result, only a very few gravimetric methods are currently used in environmental analysis.
There are four fundamental types of gravimetric analysis: physical gravimetry, thermogravimetry, precipitative gravimetric analysis, and electrodeposition. These differ in the preparation of the sample before weighing of the analyte. Physical gravimetry is the most common type used in environmental engineering. It involves the physical separation and classification of matter in environmental samples based on volatility and particle size (e.g., total suspended solids). With thermogravimetry, samples are heated and changes in sample mass are recorded. Volatile solids analysis is an important example of this type of gravimetric analysis. As the name implies, precipitative gravimetry relies on the chemical precipitation of an analyte. Its most important application in the environmental field is with the analysis of sulfite. Electrodeposition involves the electrochemical reduction of metal ions at a cathode, and simultaneous deposition of the ions on the cathode.
2. Common Procedures in Gravimetric Analysis
a. Drying to a Constant Weight
All solids have a certain affinity for water, and may absorb moisture from the laboratory air. Reagents that readily pick up water are termed hygroscopic. Those that absorb so much water that they will dissolve in it and form a concentrated solution are called deliquescent (e.g., sodium hydroxide, trichloroacetic acid). These types of substances will continually increase in weight while exposed to the air. For this reason, many types of laboratory procedures require that a sample be dried to a constant, reproducible weight (i.e., absorbed moisture removed to some standard, low level). This is especially important for the gravimetric methods. Generally, the sample is dried in a 103° C to 110° C oven for about 1 hour and allowed to cool to room temperature in a desiccator. It is then weighed, and heated again for about 30 minutes. The sample is cooled and weighed a second time. The procedure is repeated until successive weighings agree to within 0.3 mg.
b. Description and Use of the Analytical Balance
The analytical balance is the most accurate and precise instrument in an environmental laboratory. Objects of up to 100 grams may be weighed to 6 significant figures. Volumetric glassware is accurate to no more than 4 significant figures, and the accuracy of complex analytical methods rarely justifies more than 2 significant figures. Analytical balances are generally used for gravimetric analyses, and for the preparation of standard solutions. Less demanding tasks, such as the weighing of sodium phosphate for the preparation of a pH buffer, should be done using the less accurate and more robust, toploading balance.
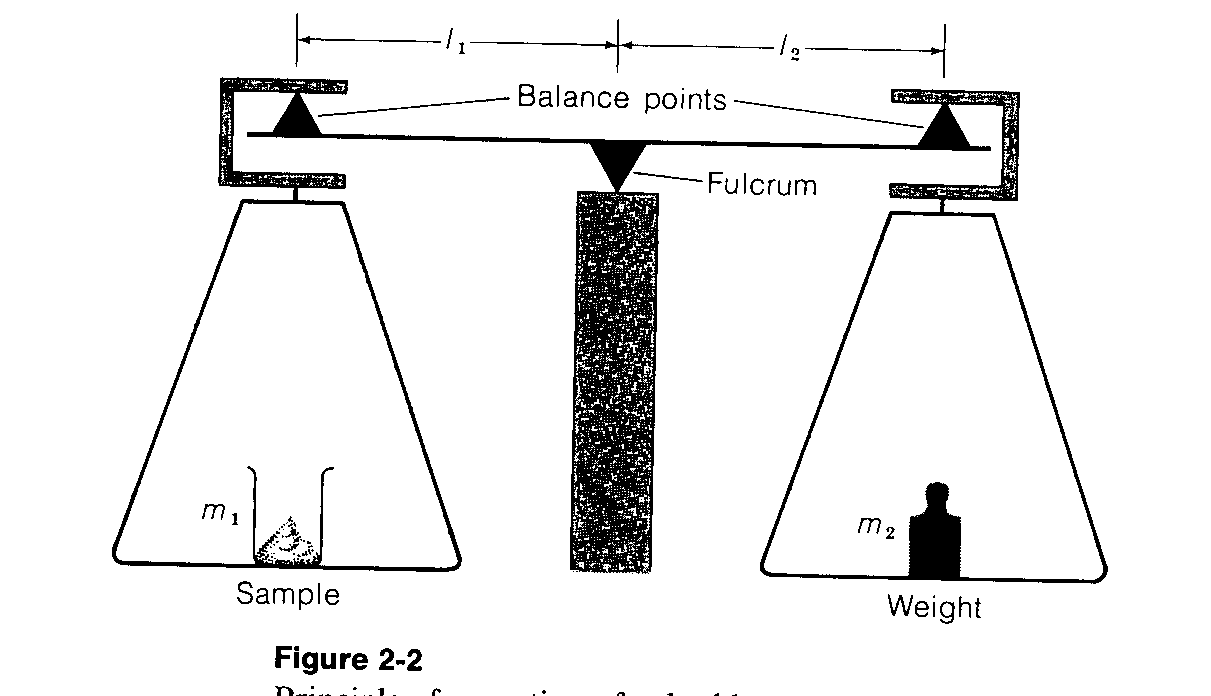
Early analytical balances were entirely mechanical with two weighing pans, one for the chemical, and one for the counterweights. Now, most analytical balances are hybrid mechanical and electronic with a single pan for the substance to be weighed. These balances use the substitution method of weighing. That is, the counterweight is fixed (hidden within the balance), and removable weights are mechanically added or subtracted from the sample side of the lever and fulcrum (see Figure 15.1).
The two pan balances are illustrated as in Figure 15.1a below. The product of the mass times the distance from the balance point to the fulcrum determines the moment about the fulcrum. When these are equal for both sample and standard (i.e., weight), the pans will be level and the balance beam horizontal.
m1l1 = m2l2
Figure 15.1a
The double pan balance
The single pan balance employs a hidden counterweight with a different fulcrum length (Figure 15.1b). This counterweight is fixed. However, weights may be added and removed from the frame of the balance pan.
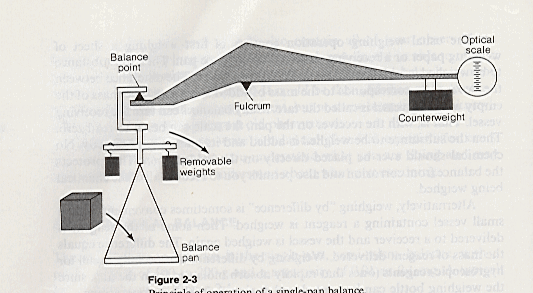
Figure 15.1b
The single-pan Balance
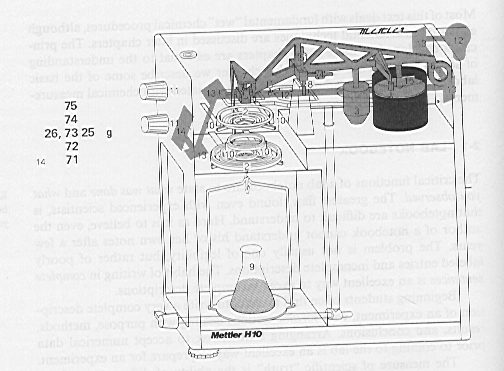
The correct weighing procedure depends somewhat on the design of the balance. Most single pan analytical balances have a knife edge supported fulcrum. Such balances are always equipped with a pan release lever which raises and lowers the fulcrum off of a finely-machined knife edge. In the Environmental Engineering Teaching Laboratory we have a Mettler H80 which employs a taught band fulcrum support system, rather than a knife edge. It, therefore, does not require a pan release. The Mettler H80 is pictured on the following page. The numbers refer to the following:
|
1. Level indicator |
8. Zero adjustment knob |
|
|
2. Weight control knob for 10 g increments |
9. Readout |
|
|
3. Weight control knob for 1 g increments |
10. Micrometer knob |
|
|
4. Power switch for optical scale |
11. Image definition adjustment |
|
|
5. Weighing pan |
12. Cover plate of lighting system |
|
|
6. Leveling feet |
13. Power cable |
|
|
7. Sliding door, right |
14. Sliding door, left |

Figure 15.2: Exterior of the Mettler H80 Analytical Balance
Some general rules should be kept in mind when using an analytical balance:
1. Allow samples to reach room temperature before weighing. Samples that are too hot will set up convection currents and the apparent sample weight will be incorrect. A slowly drifting apparent sample weight is indicative of this problem.
2. Chemicals should be placed in a weighing bottle, a plastic weighing tray, or coated weighing paper. Weighing paper is best for small quantities (usually <1g); weighing trays are used for larger amounts; and stoppered weighing bottles are recommended for reactive, volatile, toxic or hygroscopic substances. In the absence of a weighing bottle, a small beaker and watch glass may be substituted. Never place chemicals directly on the pan!
3. The analytical balance should be kept clean at all times. If a solid is spilled inside the weighing chamber, carefully remove it with the balance brush. The desiccant should be replaced or regenerated when it changes color. Used weighing papers and trays should be immediately discarded.
The procedure for weighing with the Mettler H80 is as follows:
1. Insure that the balance is level by verifying that the air bubble is in the center of the circular marking of the level indicator (1).
2. Verify that the weight control knobs (2,3) and micrometer knob (10) are in the zero position.
3. Carefully clean off the pan (5) with the soft brush provided.
4. Actuate power switch (4). Optical scale image appears.
5. Close both doors and adjust zero point, if necessary, with the zero adjustment knob (8).
6. Open one door and place the empty weighing vessel on the center of the pan. Its weight is referred to as the "tare", and the process of weighing it is called "taring". Objects to be weighed may be moved by hand prior to determination of the tare and after final weighing only. Should the sample container need to be moved between taring and final weighing, use either clean, dry rubber gloves (not latex), tongs, or a paper loop. Fingerprints will add significant mass to small samples.
7. Close the glass doors tightly to avoid unsettling air currents.
8. Turn weight control knob (2) until optical scale image moves through readout field, the turn knob (2) back one step.
9. Turn weight control knob (3) until optical scale moves into readout field.
10. Turn micrometer knob (10) until next scale division line is exactly in center of index fork.

Figure 15.3: Mettler H80 Readout
11. Read weight.
12. Open door and remove object from pan.
13. Return weight control knobs (2,3) and micrometer knob (10) to zero, and brush off pan and weighing chamber, if necessary.
14. Repeat steps 3-13 for weighing vessel plus sample.
Refer to the section on desiccators in Chapter XI.
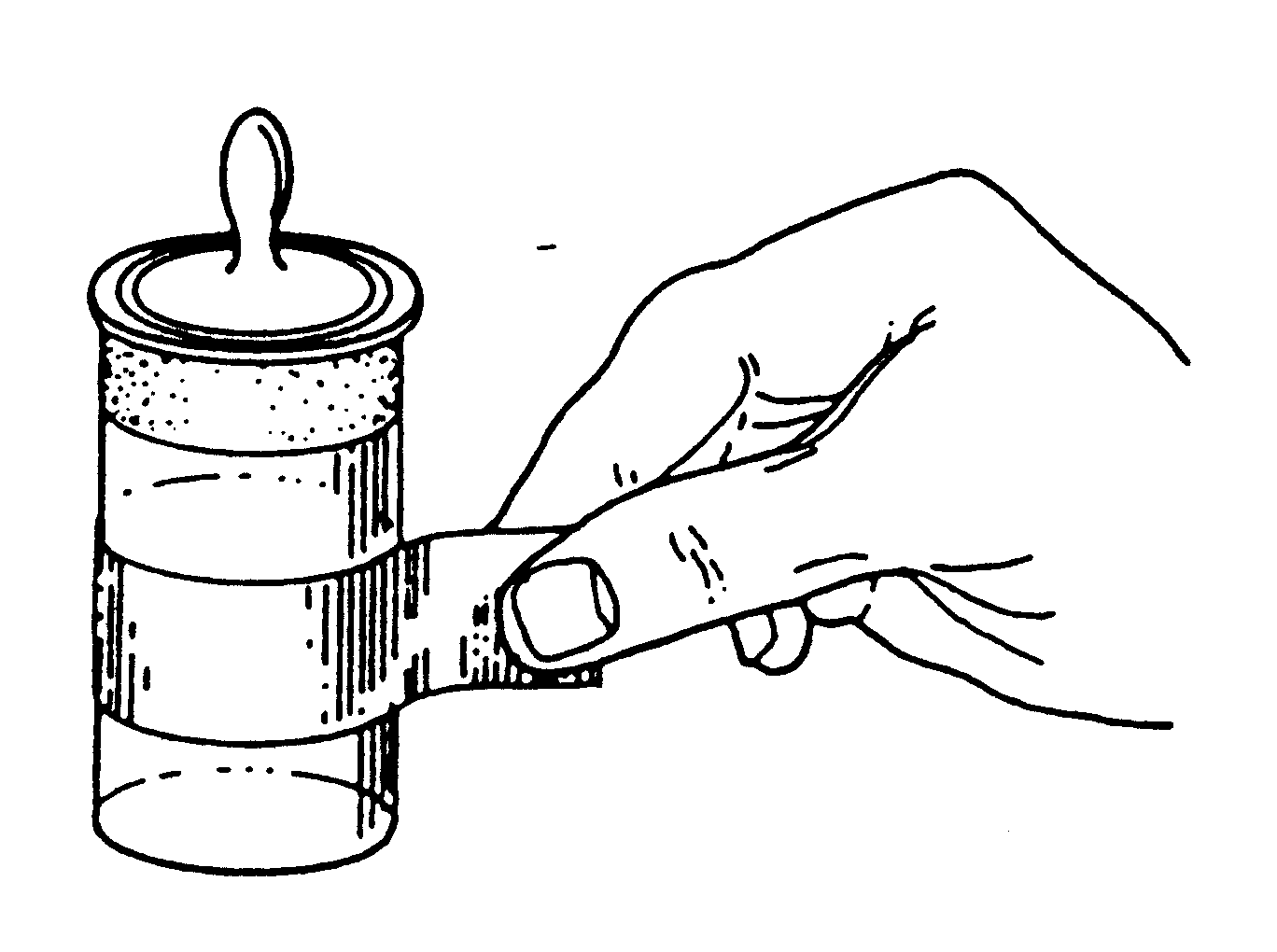
Figure 15.4 Technique for Handling a Weighing Bottle with a Paper Loop
3. Overview of Methods
A small number of gravimetric methods are in general use for the analysis of waters and wastewaters (Table 15.1). These are widely-used techniques, due largely to the tremendous importance of solids analysis in wastewater treatment.
Table 15.1
Summary of Gravimetric Methods for Environmental Analysis
|
Type |
Analyte |
Pretreatment |
|
Physical |
Total Solids |
Evaporation |
|
Suspended Solids |
Filtration |
|
|
Dissolved Solids |
Filtration + Evaporation |
|
|
Oil & Grease |
extraction with C2Cl3F3 + distillation of solvent |
|
|
Surfactants |
extraction into ethylacetate + evaporation |
|
|
Thermal |
Volatile Solids |
Evaporation + 550`C for 15 min |
|
Volatile Suspended Solids |
Filtration + 550`C for 15 min |
|
|
Precipitative |
Mg |
with Diammonium hydrogen phosphate and final pyrolysis |
|
Na |
with zinc uranyl acetate |
|
|
Silica |
precipitation/ ignition/ volatilization (with HF) |
|
|
SO4 |
with Ba |
B. PHYSICAL GRAVIMETRY
1. Total, Dissolved and Suspended Solids
a. Definitions
Total solids (TS) is generally defined as all matter in a water or wastewater sample that is not water. Because solids are not a specific chemical compound, but rather a diverse collection of dissolved and particulate matter, their concentration cannot be determined in an unambiguous way. Instead, they must be defined by the procedure used to estimated their concentration. This is referred to as an operational definition. Depending on a number of factors, the operationally defined solids concentration may be greater than, less than, or about the same as the true solids concentration. The operational definition that has been adopted through years of use is: all matter that remains as residue upon evaporation and drying at 180oC for one hour.
Total solids may be differentiated according to size into total dissolved solids (TDS) and total suspended solids (TSS; see Figure 15.7). Once again, this is an operational distinction, whereby all solids passing through filter paper of a certain pore size (e.g., 1.5 microns, Whatman #934AH) are called dissolved, and those retained are termed suspended. Therefore, the overall definition of total dissolved solids is: all matter that is not retained by a 934AH filter, and is not lost by evaporation and drying at 180oC for one hour.
b. Significance to Environmental Engineering
Most of the impurities in potable waters are in the dissolved state, principally as inorganic salts. Thus, the parameters, "total solids" and especially "total dissolved solids" are of primary importance here. Waters containing high concentrations of inorganic salts are not suitable as sources of drinking water, because such materials are often difficult to remove during treatment. Finished drinking waters containing more than 1000 mg/L TDS are generally considered unacceptable. Waters of this type may also be unsuitable for agricultural purposes due to the harmful effects of high ionic concentrations on plants. In most natural waters, the TDS (total dissolved solids) concentration correlates well with total hardness (i.e., [Ca] + [Mg]). This is useful in assessing the corrosivity of a water and the need for softening.
One may also use conductivity measurements (see Chapter XVIII) as a means of estimating the TDS (total dissolved solids) concentration. This measurement may also correlate well with the ionic strength, I. Ionic strength is numerically defined as half the sum of the product of the concentrations (in moles/liter) of each of the constituent ions times the square of their charge.
![]() (15.1)
(15.1)
Ionic strength is particularly important for predicting the chemical behavior of pollutants in water treatment and in natural systems. Russell has developed the following relationship for predicting ionic strength from conductivity measurements:
I ~ 1.6 x 10-5 x Specific Conductance (in mmho/cm) (15.2)
In a similar vein, the TDS has been used to estimate the ionic strength of natural waters via the Langlier approximation:
I ~ 2.5 x 10-5(TDS) ,where the TDS is expressed in mg/L. (15.3)
The total suspended solids (TSS) content of natural waters is of interest for the purpose of assessing particle bed load and transport. High concentrations of suspended matter may be detrimental to aquatic life. In theory, TSS could be used for assessing particle removals during water treatment. However, nearly always the concentration of colloidal particles in water is measured as turbidity since this latter technique is faster and more precise. Volatile solids determinations are rarely made on potable water samples since far more accurate and precise information on organic content can be obtained by TOC (Total Organic Carbon) analysis.
In the realm of municipal wastewater, suspended solids analysis is by far the most important gravimetric method. It is used to evaluate the strength of the raw wastewater as well as the overall efficiency of treatment. Furthermore, most WWTP's have effluent standards of 10 to 30 mg/L suspended solids which may be legally enforceable. In particular, the volatile suspended solids is used in activated sludge to estimate the standing biomass in mixed liquor. In this type of application, the additional time and expense associated with more direct organic measurements such as TOC is not warranted. Total and volatile solids analyses are used in the design and operation of sludge digestors, vacuum-filters and incinerators.
As was the case with municipal wastewater, suspended solids analysis is useful as a means of assessing the strength of industrial wastewaters and the efficiency of industrial wastewater treatment. Since some industrial wastes contain unusually high concentrations of inorganic ions, the total solids and conductivity tests can be useful for determining the amenability of such waters to anaerobic treatment.
Table 15.2
Applications of Solids Analysis in Environmental Engineering
|
Application |
TS |
VS |
TDS |
TSS |
VSS |
|
Drinking Water |
X |
||||
|
Natural Waters |
X |
X |
|||
|
Municipal Wastewater |
X |
X |
|||
|
Industrial Wastewater |
X |
X |
X |
X |
X |
|
Sludge |
X |
X |
Table 15.3
Some Typical Solids Concentrations
|
Source |
Concentration (mg/L) |
|||||
|
Low |
Avg |
High |
||||
|
NATURAL WATERS |
||||||
|
Fresh |
TDS |
20 |
120 |
1,000 |
||
|
Brines |
TDS |
5,000 |
300,000 |
|||
|
DOMESTIC WASTEWATER |
||||||
|
Raw |
TDS |
350 |
600 |
900 |
||
|
VDS |
165 |
285 |
600 |
|||
|
TSS |
100 |
200 |
350 |
|||
|
VSS |
75 |
135 |
215 |
|||
|
Secondary Effluent |
TSS |
10 |
30 |
60 |
||
|
Activated Sludge Mixed Liquor (conventional) |
TSS |
1,500 |
3,000 |
|||
|
Activated Sludge Mixed Liquor (extended aeration) |
TSS |
3,000 |
6,000 |
|||
|
Primary Sludge |
TSS |
20,000 |
70,000 |
|||
|
Secondary Sludge |
TSS |
5,000 |
12,000 |
|||
|
STORM WATER |
TSS |
5 |
300 |
3,000 |
||
2. Sampling
Sampling for Solids Analysis. Suspended solids present a special sampling problem. In order to avoid unrepresentative sampling or particle break-up, it is important that: (1) the fluid stream to be sampled is completely mixed at the point of sampling; and (2) the disturbance of the flow pattern of this stream is minimized, especially at the mouth of the sampling device. This means that sample intake velocities should be similar to the flow velocities at the point of mixing. Figure 15.5 shows the percent error expected at different ratios of intake velocity to stream velocity, and as a function of particle size. Note that intake velocity is less important for smaller particles. Finally, the sampler intake should face into the stream flow (within 20 degrees).
The possible presence of settleable solids may also require that some type of depth-integrated sample be collected. If it can be demonstrated that mixing is sufficient at the point of sampling to produce a homogeneous cross-section of settleable solids, depth-integrated sampling should not be necessary. Laboratory subsampling (removing a representative aliquot from a sample in the laboratory) is best accomplished by shaking, then rapidly pouring a portion of the sample into a receiving vessel (see Figure 15.6). Use as large a sample aliquot as possible. A good general reference on sampling techniques is the document: Handbook for Sampling and Sample Preservation of Water and Wastewater (USEPA, 1982). For an excellent presentation on sampling suspended solids in natural aquatic systems see: Guy & Norman (undated).
Samples should be collected in clean glass or high density polyethylene. Analysis should be conducted as soon after sampling as possible. Highly alkaline waters will leach metals from glass over time. Special care should be taken with waters high in manganese or iron. If these water are in contact with air or dissolved oxygen for extended periods of time the dissolved metals may become oxidized and precipitate.
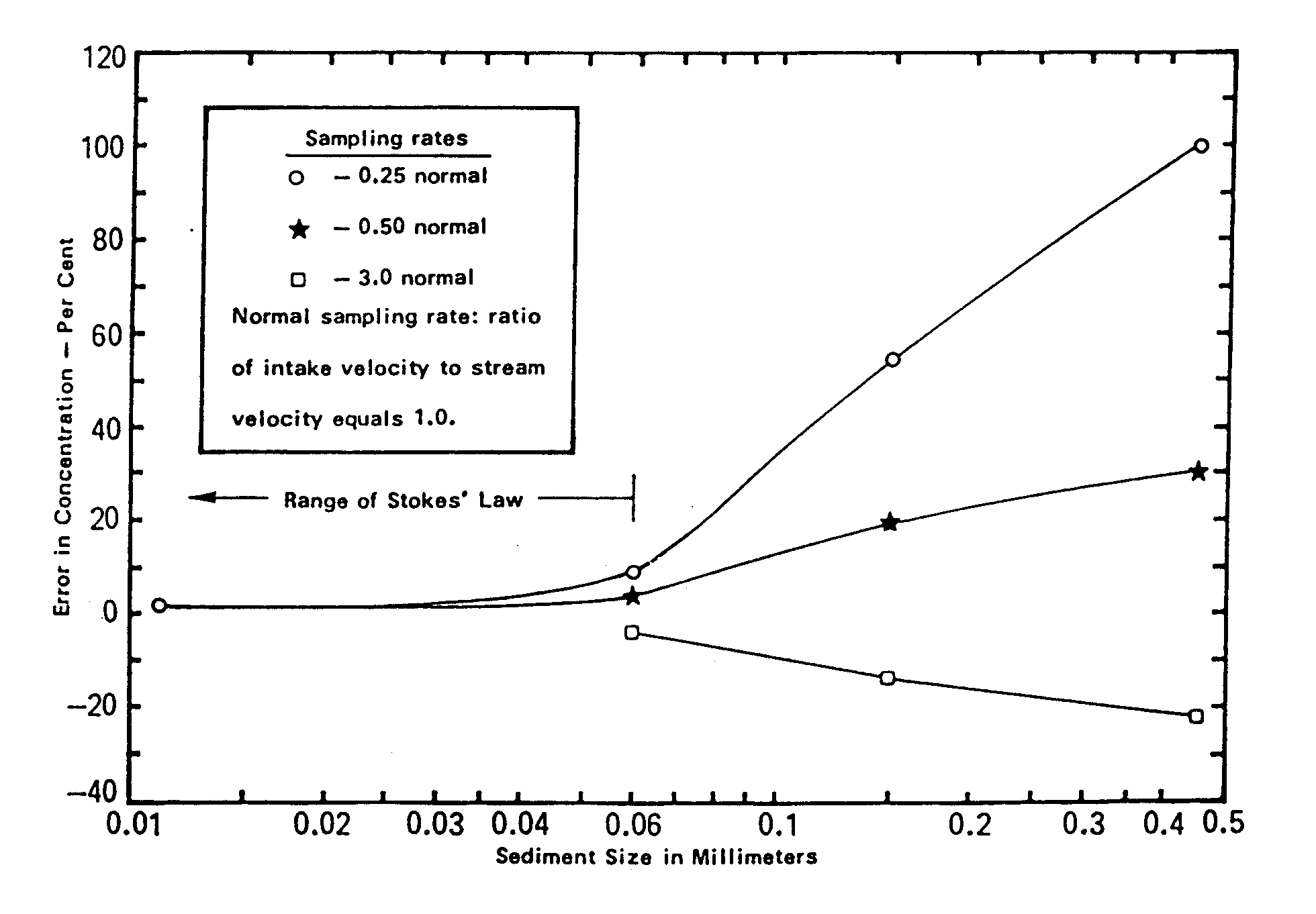
Figure 15.5: Relationship Between Sediment Size and Sampling Bias
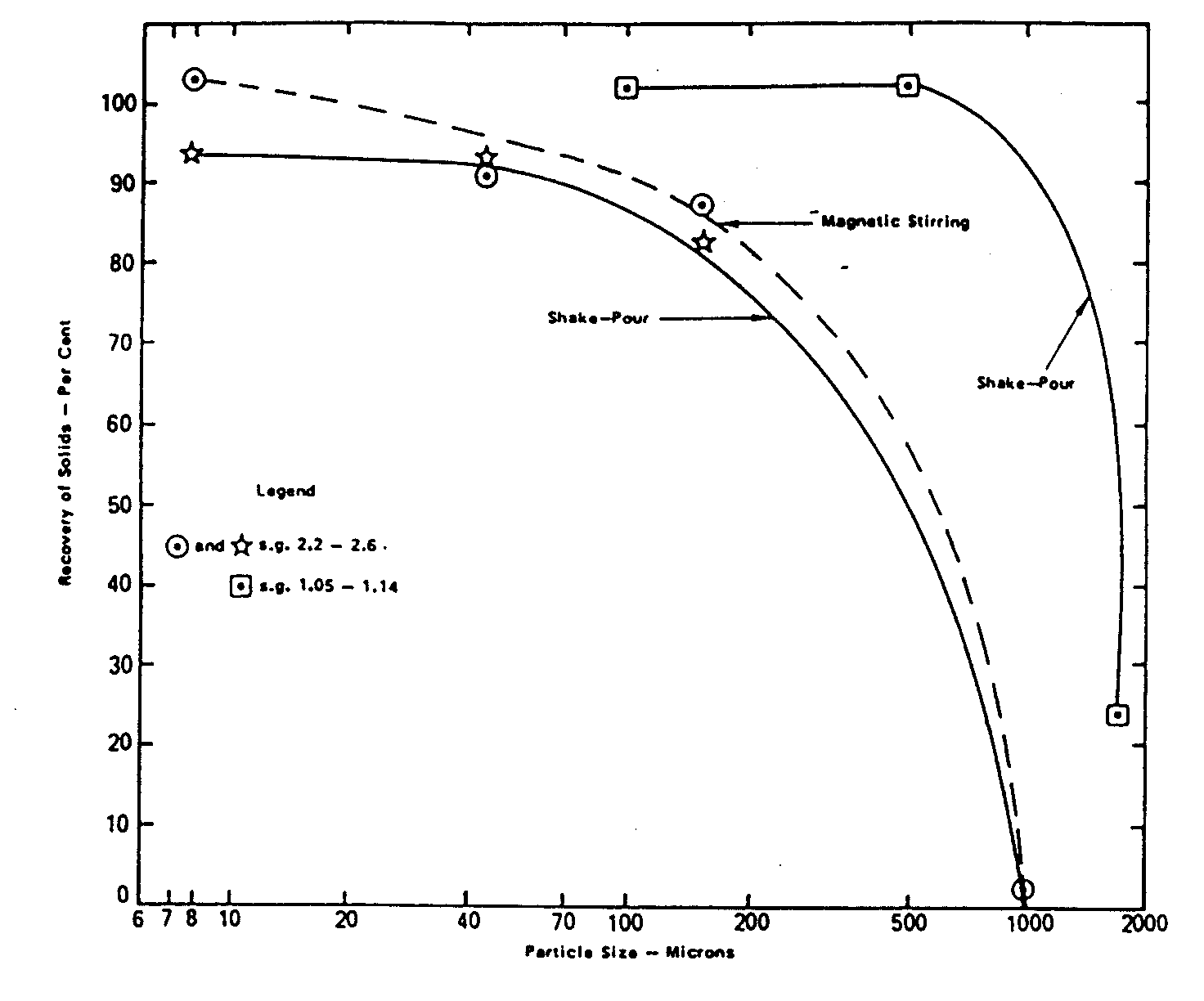
Figure 15.6
Recovery vs Particle Size During Subsampling with Different Mixing Techniques
Figure 15.7
Fractionation of Solids in Analysis of Water and Wastewater
3. Procedures
Total Solids (Total Residue). Total solids is determined by the final weight of a dried sample (minus tare) divided by the original sample volume. Evaporating dishes of platinum, vycor or porcelain may be used. Platinum is preferred, because it is more inert than the other two, and can be heated to a constant weight more easily. However, platinum is very expensive, so vycor is often used. Porcelain is difficult to bring to a constant weight, and its use should be avoided. Space permitting, evaporating dishes should be stored in a desiccator so as to avoid the collection of dust and absorption of moisture while not in use. The precision of this method has been estimated to be ± 4 mg or ± 5%. However, settled wastewater may give better precision, on the order of ± 1 mg.
1. Preheat a 100 mL evaporating dish at 550± 50° C for 1 hour, cool in a in a drying oven or in the open air (protected from dust) for 15-20 minutes, bring to room temperature in a desiccator, and weigh. Repeat until a constant weight is achieved.
2. Measure 75 mL of sample or a volume sufficient to yield 200 mg TS, whichever is less. Add this to the preweighed dish and evaporate to dryness in a drying oven set at 98° C. Alternatively, a steam bath may be used.
3. Dry for an additional hour at 103-105° C.
4. Cool in a desiccator and weigh. As always, the sample should be re-heated in accordance with #3 and re-weighed to achieve a constant weight.
Dissolved Solids (Filtrable Residue). Dissolved solids may be determined directly by analysis of the filtered sample for total solids, or indirectly by determining the suspended solids and subtracting this value from the total solids. When using the direct method, final drying may be conducted at one of two temperatures. Although solids measurements are operationally defined, and are therefore inherently without bias, one can postulate a method accuracy based on the theoretical solids definition (all that is not water). Using this as a basis of comparison, the choice of drying temperatures can have an important effect on accuracy. At 103-105° C positive bias may result from physically occluded water and the retention of waters of crystalization. This is especially true for waters high in calcium, magnesium, chloride or sulfate. The problem of physically occluded water may be exacerbated by the presence of large amounts of residue (i.e., more than 200 mg). On the other hand, some conversion of bicarbonate to carbon dioxide and subsequent volatilization may cause a small amount of negative bias. At 180± 2° C occluded water is removed, but some water of crystallation may still remain, especially in the presence of sulfates. Furthermore, at this higher temperature additional volatilization of certain inorganic salts (e.g., carbonate, some chlorides and nitrates) will occur. Overall, drying to 180± 2° C seems to give results for TDS that are closest to what one would calculate from a complete chemical analysis. For this reason, it is included as an option for the determination of TDS. In general, either temperature may be used for waters of low color and low alkalinity. Hard waters, alkaline waters, or highly-colored waters should probably be dried at the higher temperature.
1. Prewash a 934AH glass fiber filter by rinsing three times with 20 mL of distilled water. Maintain suction until the filter is dry.
2. Analyze the filtrate in accordance with the total solids procedure. Final drying (1 hour period) may be conducted at either 103-105° C or 180± 2° C.
Suspended Solids (Non-filtrable Residue). Suspended solids is measured directly by drying and weighing the solids retained during filtration. This approach is much more accurate for most waters than the indirect method of subtracting dissolved solids from total solids. Whatman 934AH glass fiber filters (nominal pore size 1.4 microns) are most commonly used. These may be placed in small aluminum foil weighing pans. The filters may be identified by etching a number into the small tab on the weighing pans. Filters should be weighed, heated and dried while remaining in the aluminum pans. They should only be removed for actual filtering operations. They should always be handled with tongs, and one must be careful not to tear them or leave fragments on the filtration apparatus. Precision has been found to be ± 5 mg/L at low concentrations, increasing up to ± 20 mg/L at values of 200 mg/L or more.
1. Prewash a 934AH glass fiber filter by rinsing three times with 20 mL of distilled water. Maintain suction until the filter is dry.
2. Dry this filter at 103-105° C for 1 hour, and cool in a desiccator.
3. Weigh the filter, then pass a water sample of sufficient volume to yield 50-200 mg suspended solids through it. Smaller volumes will result in reduced accuracy.
4. Dry for at least one hour at 103-105° C.
5. Cool in a desiccator and weigh. As always, the sample should be re-heated in accordance with #4 and re-weighed to achieve a constant weight (a drop of no more than 0.5 mg).
C. THERMOGRAVIMETRY AND COMBUSTION ANALYSIS
Thermogravimetry and combustion analysis involve the heating of a sample to 500° C or more with the oxidation and/or volatilization of some of the sample constituents. Either the change of sample weight is determined (thermogravimetry), or the combustion gases are trapped and weighed (combustion analysis). The former method is used for volatile solids analysis in environmental engineering. The latter is used in many fields of science for the determination of total carbon and hydrogen in solids.
With thermogravimetric methods, it is especially important to return the sample to room temperature before weighing. Otherwise the differences in temperature will create convection currents around the balance pan, which will severely disrupt method accuracy. A steady increase in apparent weight while the sample is on the pan indicates a problem of this type. Large vessels and samples will require longer cooling times to dissipate their excess heat.
1. Volatile Solids and Fixed Solids
Fixed solids are those that remain as residue after ignition at 550° C for 15 minutes. The weight of material lost is called the volatile solids. Thus the total operational definition for volatile solids would be: all matter lost upon ignition at 550° C for 15 minutes, but not lost upon drying at 103-105° C for 1 hour. The portion lost upon ignition is generally assumed to be equivalent to the organic fraction. The portion remaining is considered the inorganic fraction. For waters of moderate to high hardness, most of this is calcium carbonate which decomposes only at temperatures exceeding 800° C. When igniting a filter with suspended matter, one must be especially careful of the temperature; above 600° C glass fiber filters begin to melt and can loose a significant amount of weight in 15 minutes.
Combining the fractionations resulting from ignition and filtration, one arrives at a total of 9 separate categories: total solids (TS), fixed solids, volatile solids, total dissolved solids (TDS), fixed dissolved solids, volatile dissolved solids, total suspended solids (TSS), fixed suspended solids, and volatile suspended solids (VSS). In practice, only four of these (TS, TDS, TSS, and VSS) are commonly used. When comparing fixed solids with inorganic content, one would expect positive bias from incomplete oxidation of organic matter, and negative bias from decomposition of certain inorganics. Ammonium salts may be lost during low temperature drying or upon ignition. Most others are stable under the conditions used for volatile solids determination with the exception of magnesium carbonate (equation 15.4). Volatile solids may be effected by these as well as loss of recalcitrant waters of crystallation (positive bias), and previous losses of organic matter to volatilization during low-temperature drying (negative bias). A modest interlaboratory study found an average standard deviation of 11 mg/L on a sample of 170 mg/L volatile solids.
MgCO3 ------------> MgO + CO2 (15.4)
Analysis of sludge, sediment, and soil for fixed and volatile solids presents additional problems. Both negative and positive biases can be acute, so greater care must be taken in ignition temperature and time. Again, ammonium compounds (often present in sludge in the form of ammonium bicarbonate) may be lost during low temperature drying and therefore should not introduce a bias in volatile solids analysis (equation 15.5). However, occluded water can be especially problematic. Standard practice is to use a 1-hour ignition time. With sludge, sediment and soil, nearly all of the solids are in the undissolved state. Therefore, only total, total volatile, and total fixed fractions are normally determined.
NH4HCO3 ---------> NH3 + H2O + CO2 (15.5)
1. Dry and weigh a vessel containing the solids to be analyzed. For volatile and fixed suspended solids analysis, the filter (with residue) prepared for suspended solids analysis and dried to a constant weight may be used. For volatile and fixed total (or dissolved) solids, the evaporating dish (with residue) prepared for total (or dissolved) solids analysis dried to a constant weight should be used.
2. Ignite the sample and vessel in a preheated muffle furnace set at 550± 50° C for 15-20 min (water & wastewater) or 1 hour (sludge, sediment & soil).
3. Cool for 15 minutes in the open air in an area protected from dust.
4. Place vessel in a desiccator for final cooling to room temperature and weigh. Due to the approximate nature of this test samples are not generally re-heated and dried to a constant weight.
2. Carbon & Hydrogen Analysis
Total carbon and hydrogen can be determined in a solid by combustion in the presence of pure, dry oxygen (Figure 15.8). The combustion gases are oxidized completely to CO2 and H2O with the aid of a transition metal catalyst. The first trap, phosphorus pentoxide (P2O5), efficiently removes all water formed. Recall that P2O5 is deliquescent, and an efficient desiccant (Table 11.2). The second contains ascarite, which is sodium hydroxide-impregnated asbestos. This will trap all of the carbon dioxide (equation 15.6). The third tube protects the other two from backflow of atmospheric water and carbon dioxide. After the sample is completely oxidized, the tubes are weighed to determine the increase. This type of analysis is sometimes conducted on dried sludges, sediments, soils, and extracted aquatic organic matter.
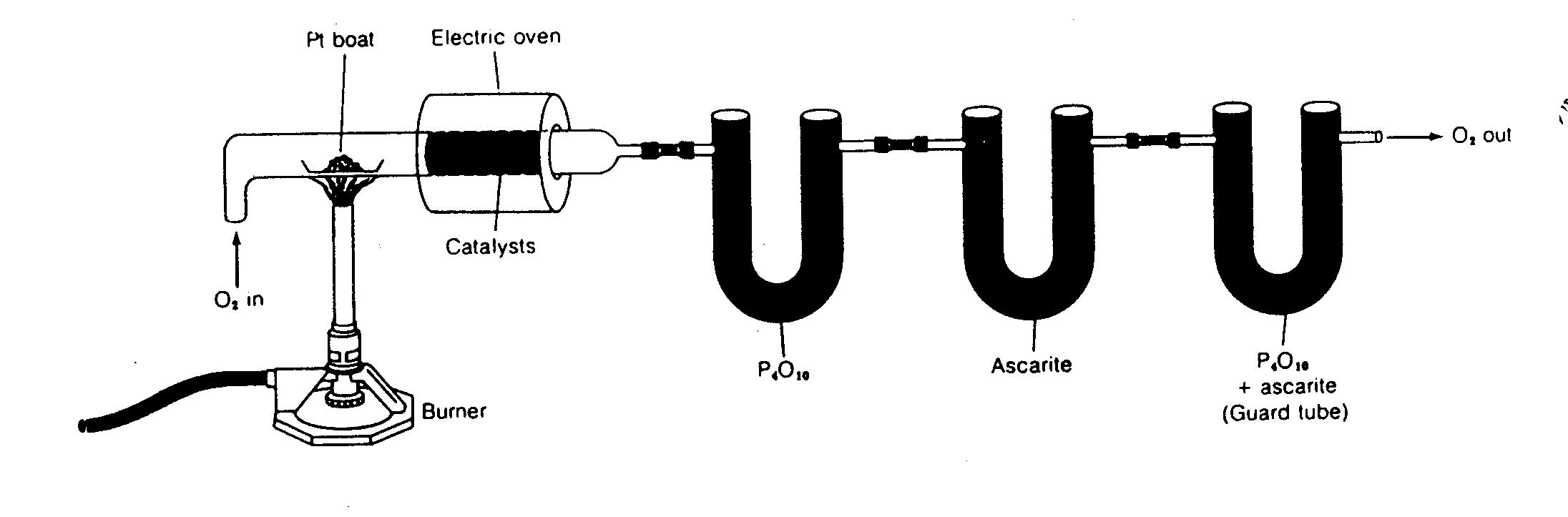
Figure 15.8
Thermogravimetric Analysis for Carbon and Hydrogen
NaOH + CO2 ------------> NaHCO3 (15.6)
D. PRECIPITATIVE GRAVIMETRIC ANALYSIS
Precipitative gravimetric analysis requires that the substance to be weighed be readily removed by filtration. In order for a non-filtrable precipitate to form, it must be supersaturated with respect to its solubility product constant. However, if it is too far above the saturation limit, crystal nucleation may occur at a rate faster than crystal growth (the addition of molecules to a crystal nucleus, eventually forming a non-filtrable crystal). When this occurs, numerous tiny micro-crystals are formed rather than a few large ones. In the extreme case, micro-crystals may behave as colloids and pass through a fibrous filter. To avoid this, precipitating solutions may be heated. Because the solubility of most salts increases with increasing temperature, this treatment will lower the relative degree of supersaturation and slow the rate of nucleation. Also, one might add the precipitant slowly with rapid mixing to avoid the occurrence of locally high concentrations.
Precipitative gravimetry is often practiced at high ionic strengths. This is to reduce the electric double layer thickness (salting-out effect) of the slowly forming crystals. When this occurs, electrostatic repulsion between the crystal and its precipitating molecules is reduced. Crystal growth can then occur more rapidly.
It is very important that the precipitate be pure and have the correct stoichiometry. Coprecipitation occurs when an unwanted ion or molecule becomes trapped in the precipitate. This may be due to inclusion or occlusion. Inclusion is the term used of a single subsitution in the crystal lattice by an ion of similar size. Occlusion refers to the physical trapping of a large pocket of impurities within the crystal. One technique for minimizing these problems is to remove the mother liquor, re-dissolve the precipitate and then re-precipitate. The second time the mother liquor will contain fewer unwanted ions capable of coprecipitation.
1. Sulfate
The method of choice for sulfate in waters and wastewaters is the precipitative gravimetric procedure using barium (APHA et al., 1985). If Ba(+II) is added in excess under acidic conditions, BaSO4 is precipitated quantitatively. The reaction is allowed to continue for 2 hours or more at 80-90oC. This is to encourage the formation of BaSO4 crystals (non-filtrable) from the initially formed colloidal precipitate (partially filtrable). The precipitate is washed, and then dried at 800`C for 1 hour. Low pH is needed to avoid the precipitation of BaCO3 and Ba3(PO4)2. Positive bias may result from acid-resistant insoluble matter such as silica, sulfites which may oxidize to sulfate, and nitrate and chloride which will associate with barium and co-precipitate to a small extent with the barium sulfate. Negative bias may result from the presence of certain heavy metals (e.g., Cr, Fe) which can form soluble complexes with sulfate.
Ba+2 + SO4-2 = BaSO4(ppt.) (15.7)
Chloride may be determined by precipitation with silver. Interfering ions likely to form insoluble silver salts are the other halogens (bromide, iodide), cyanide, and reduced sulfur species (sulfite, sulfide, and thiosulfate). Fortunately, the reduced sulfur compounds can be pre-oxidized with hydrogen peroxide, and the others are rarely present at high concentrations. Although AgCl can be determined gravimetrically, the recommended procedure for water and wastewater (APHA et al., 1985) is to use a volumetric procedure with chromate as an indicator (ppt. of red silver chromate; see Chapter 16).
E. ELECTRODEPOSITION
Electrodeposition may be thought of as a special case of precipitative gravimetry where electrons are the precipitant. It is generally used for the determination of metals in concentrated solution. However, it has few, if any applications in environmental engineering analysis.
REFERENCES
APHA, AWWA, WPCF (1985) Standard Methods for the Examination of Water and Wastewater, APHA, Washington, 16th edition, pp. 32-34, 76-80, 92-100.; or 15th edition, pp. 30-32, 70-73, 90-98.; or 14th edition, pp. 34-36, 71-75, 89-98.
Guy, H.P. & Norman V.W., Techniques of Water-Resources Investigations of the United States Geological Survey, Book 3, Chapter C2, Field Methods for Measurement of Fluvial Sediment.
Hem, J.D., Chapt. 4 in Water Analysis, Volume 1, Minear & Keith eds.
Hem, J.D., USGS Water Supply Paper #1473: 1st ed. 1959; 2nd ed. 1970.
Rubinson, K.A. (1987) Chemical Analysis, Little, Brown & Co., Publ., Boston., pp. 178-179, 243-245.
Sawyer, C.N. & P.L. McCarty (1978) Chemistry for Environmental Engineers, McGraw Hill Publ,. pp. 65-69, 284-285, 454-462.
Snoeyink, V.L. & Jenkins, D. (1980) Water Chemistry, John Wiley & Sons, Publ., New York, pp. 74-82.
USEPA (1982) Handbook for Sampling and Sample Preservation of Water and Wastewater, US EPA, EMSL, September, 1982; EPA-600/4-82-029 [Gov.Docs.: EP1.23/5:600/4-82-029].
PROBLEMS
15.1 Would you expect a positive bias, a negative bias, or no bias from the following errors or difficulties in solids analysis? Why?
|
a. use of a coarse filter rather than a 934AH in TSS analysis. |
|
|
b. use of a top loading balance rather than an analytical balance for TDS analysis. |
|
|
c. insufficient cool-down time for TDS analysis. |
|
|
d. excessively high ignition temperature (650oC) for VSS analysis. |
|
|
e. incomplete drying of a filter prior to determining its tare in TSS analysis |
|
|
f. clogging of a filter in TSS analysis. |
15.2 Five hundred mL of a river water are subjected to sulfate determination by the barium gravimetric method. The final weight of the precipitate was 73.5 mg. What was the sulfate concentration in mg/L?